Clear Sky Science · sv
Statistisk kristallografi avslöjar ett allostert nätverk i SARS-CoV-2 Mpro
Hur proteins formskiftande hjälper ett coronavirus-enzym att fungera
Viruset som orsakar COVID-19 är beroende av en molekylär maskin, kallad huvudproteaset, för att klippa långa virala kedjor till fungerande delar. Läkemedel riktar sig redan mot detta enzym, men forskare har fortfarande svårt att se hur dess subtila interna rörelser styr när det slås på eller av. Denna studie använder mer än tusen ögonblicksbilder av proteaset låst i kristaller för att avslöja hur avlägsna delar av molekylen kommunicerar med den aktiva delen där kemin sker — och visar hur denna dolda kommunikation kan störas.
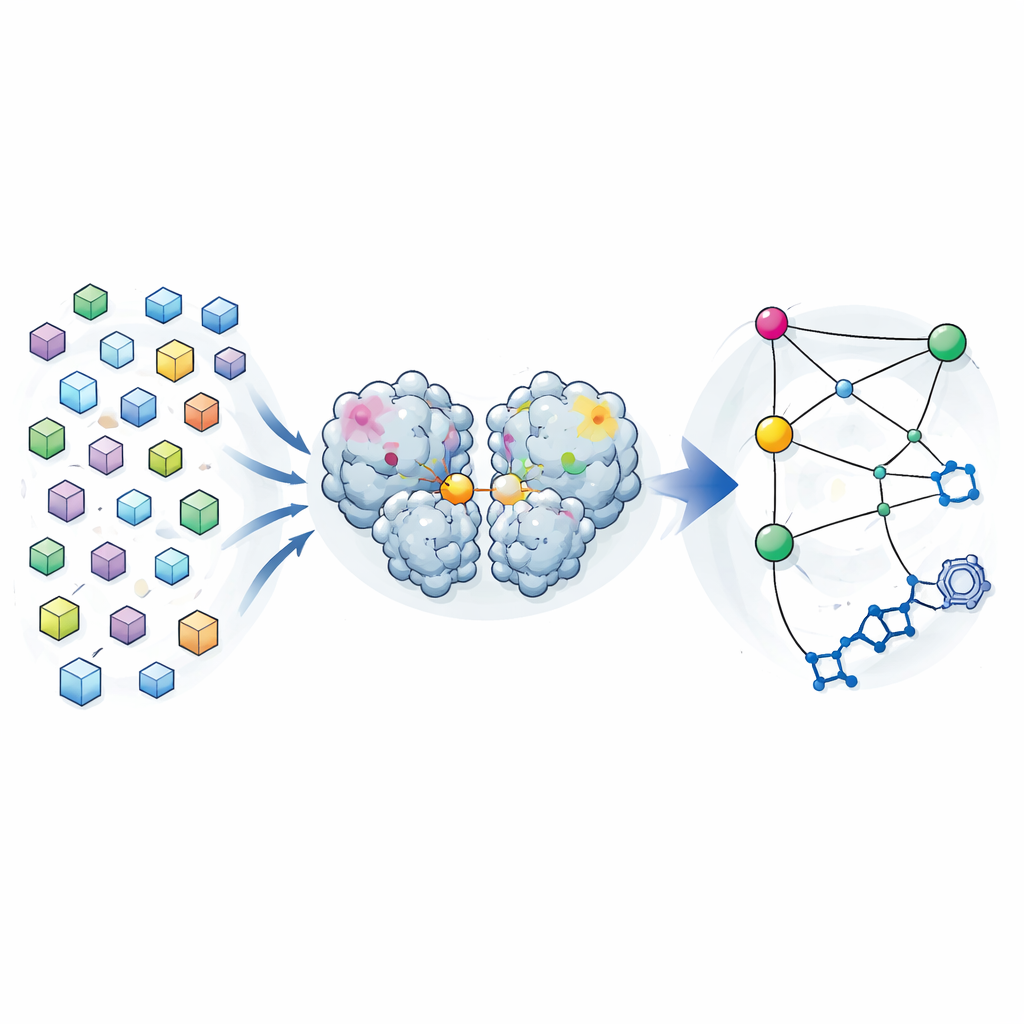
Många ögonblicksbilder istället för bara en
Traditionell kristallografi syftar till att ge en enskild ”bäst” struktur av ett protein, genomsnittligt över många molekyler i en kristall. Författarna vände på denna logik. Under en tidigare läkemedelssökningskampanj hade de samlat diffraktionsdata från nästan 7000 kristaller av SARS-CoV-2:s huvudproteas. Efter noggrann filtrering av kristaller som innehöll bundna föreningar och tillämpning av strikta kvalitetsgränser återstod 1146 högupplösta strukturer av enzymet ensamt. Istället för att betrakta små skillnader mellan dessa strukturer som ren brus analyserade de dem som en fördelning av former som proteaset kan anta samtidigt som det förblir nära sin fungerande form.
Hitta långdistanskopplingar inne i enzymet
Genom att jämföra hur varje atoms position varierade från struktur till struktur byggde teamet en kovarianskarta — en slags statistiskt fingeravtryck som visar vilka regioner som tenderar att röra sig tillsammans. De fann att dessa kristallbaserade korrelationer starkt liknade dem som förutsagts av långa molekylära dynamiksimuleringar av proteaset i lösning. Det antydde att den variabilitet som frysts in i kristallerna återspeglar verkliga flexrörelser som enzymet utforskar i vatten. När de fokuserade på hur olika regioner korrelerade med den aktiva ytan — fickan som utför klippningen — upptäckte de tre ”hotspots” i proteasets dimeriseringsdomän, den del som hjälper två kopior av enzymet att para ihop sig. Förändringar vid dessa hotspots, långt från den aktiva ytan, verkade vara starkt kopplade till förändringar i den katalytiska fickan.
Testa hotspots med riktade förändringar
För att undersöka om dessa statistiskt identifierade platser verkligen styr funktionen ändrade forskarna varje hotspot genom att ersätta en enda aminosyra med alanin, ett standard sätt att knuffa lokal struktur. De mätte sedan hur väl varje mutantproteas bildade dimerer, bundet ett substratlikt fragment och klippte ett fluorescerande testpeptid. En position, N214, uppträdde som en bekräftad ”alloster hotspot”: mutationen försvagade dimerbildningen mer än tiofaldigt och även när tillräckligt många dimerer fanns sjönk katalyshastigheten med ungefär en storleksordning. Högupplösta strukturer av mutanten visade varför: det lokala vätebindningsnätverk som normalt stabiliserar en avgörande slinga som formar ”oxyanjonhålet” i den aktiva ytan bröts, och denna slinga blev delvis oordnad och felaktigt inriktad för kemi.
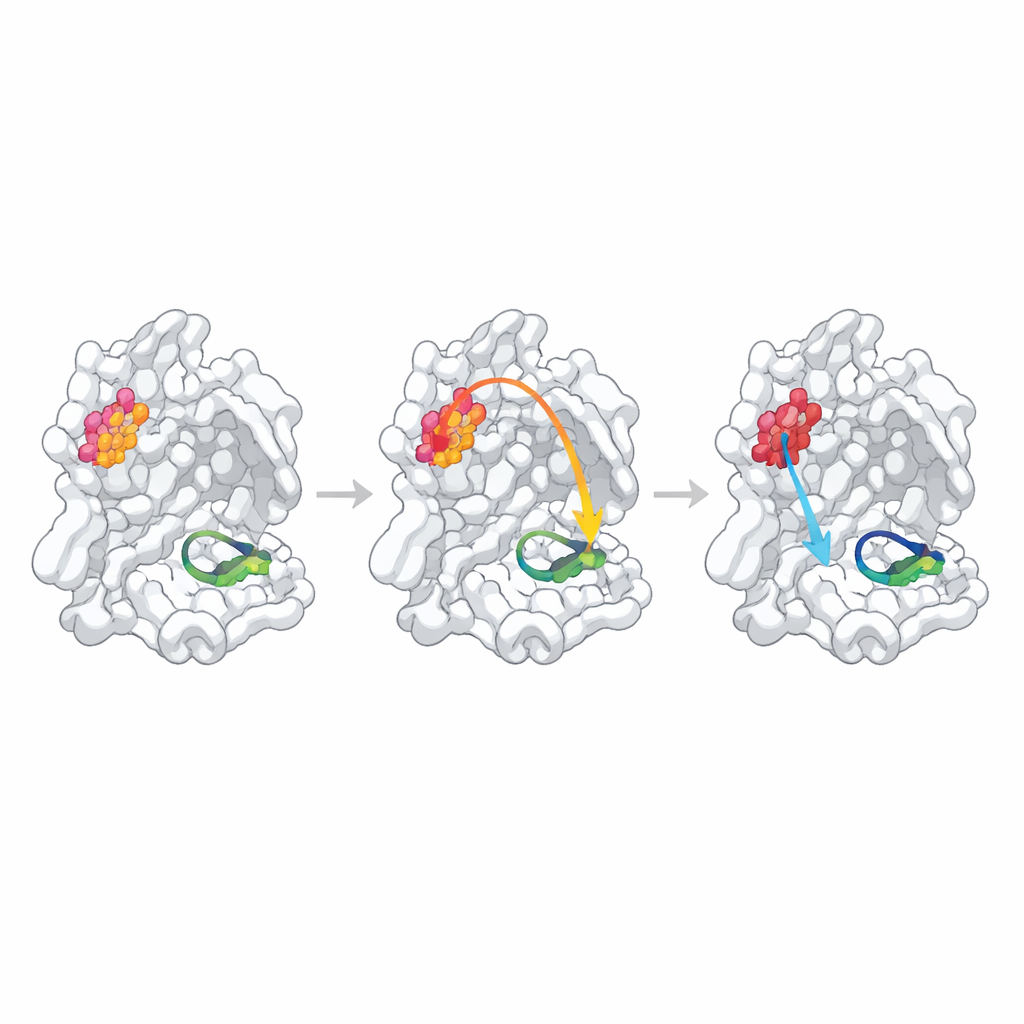
När förutsagda länkar är svaga eller dolda
De andra två förutsagda hotspots uppträdde annorlunda. Att byta Q256, en rest exponerad för lösningsmedel och inte uppenbart involverad i dimerkontakter, gav bara en måttlig minskning av aktiviteten som i stort sett kunde förklaras av något svagare dimerbildning och något sämre bindning av testinhibitorn. Dess kristallstruktur avslöjade en helt ny packningsarrangemang där närliggande molekyler fyllde håligheten som lämnats av mutationen, vilket gjorde det svårt att skilja verklig intern kommunikation från kristallspecifika effekter. Den tredje platsen, S284, ligger i en liten ”blixtlås” som förbinder de två enzymkopiorna. Som förväntat från sekvensjämförelser med närbesläktade coronavirus hade ersättningen av denna rest nästan ingen effekt på struktur eller funktion, vilket tyder på att denna särskilda alaninändring var en för mild störning för att testa den förutsagda vägen.
Kristaller som små kraftexperiment
Ett intressant sidoresultat av arbetet är att många av strukturvariationerna mellan kristaller kunde spåras till små förändringar i kristallgittret — den upprepade arrangemangen av proteiner och lösningsmedel. När kristaller torkade i något olika grad under hantering krympte eller sträcktes deras enhetsceller, och dessa skift ryckte i kontaktpunkterna mellan intilliggande proteasmolekyler. De krafterna spreds genom proteinet och genererade den strukturella mångfald som låg till grund för kovariansanalysen. Med andra ord agerade varje kristall som ett litet mekaniskt experiment, varsamt spännande proteaset i olika riktningar och avslöjande hur det böjer sig utan att falla isär.
Varför detta är viktigt för biologi och läkemedelsdesign
Genom att behandla tusentals kristallstrukturer som en statistisk population snarare än ett enda svar kunde författarna härleda ett allostert nätverk som länkar en dimergränsrest, N214, till det katalytiska centret i SARS-CoV-2:s huvudproteas. Att störa denna plats minskar inte bara hur många dimerer som bildas; det rör specifikt till geometrin i den aktiva slingans konfiguration som krävs för effektiv katalys. Detta bekräftar att enzymets aktivitet styrs av långdistanskommunikation över proteinet. Mer allmänt demonstrerar studien ett "statistisk kristallografi"-angreppssätt där okontrollerade, vardagliga störningar av kristaller — såsom små skillnader i uttorkning — blir en resurs för att kartlägga funktionella rörelser. När liknande dataset samlas in för andra proteiner kan denna strategi hjälpa till att avslöja nya reglerande platser och vägleda designen av läkemedel som inte bara fungerar genom att blockera aktiva ytor utan genom att omkoppla de interna samtal som får proteiner att fungera.
Citering: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Nyckelord: SARS-CoV-2 huvudproteas, allosteri, proteindynamik, kristallografi, läkemedelsupptäckt