Clear Sky Science · pl
Kryształografia statystyczna ujawnia sieć allosteryczną w Mpro SARS-CoV-2
Jak zmiany kształtu białka pomagają enzymowi koronawirusa działać
Wirus wywołujący COVID-19 polega na maszynie molekularnej zwanej główną proteazą, która tnie długie łańcuchy wirusowe na funkcjonalne fragmenty. Ten enzym jest już celem leków, ale naukowcy wciąż mają trudności ze zrozumieniem, jak subtelne wewnętrzne ruchy kontrolują jego przełączanie między stanem aktywnym a nieaktywnym. Badanie wykorzystuje ponad tysiąc „migawek” proteazy zamrożonej w kryształach, aby ujawnić, jak odległe części cząsteczki komunikują się z miejscem katalitycznym — i pokazuje, jak tę ukrytą komunikację można zaburzyć.
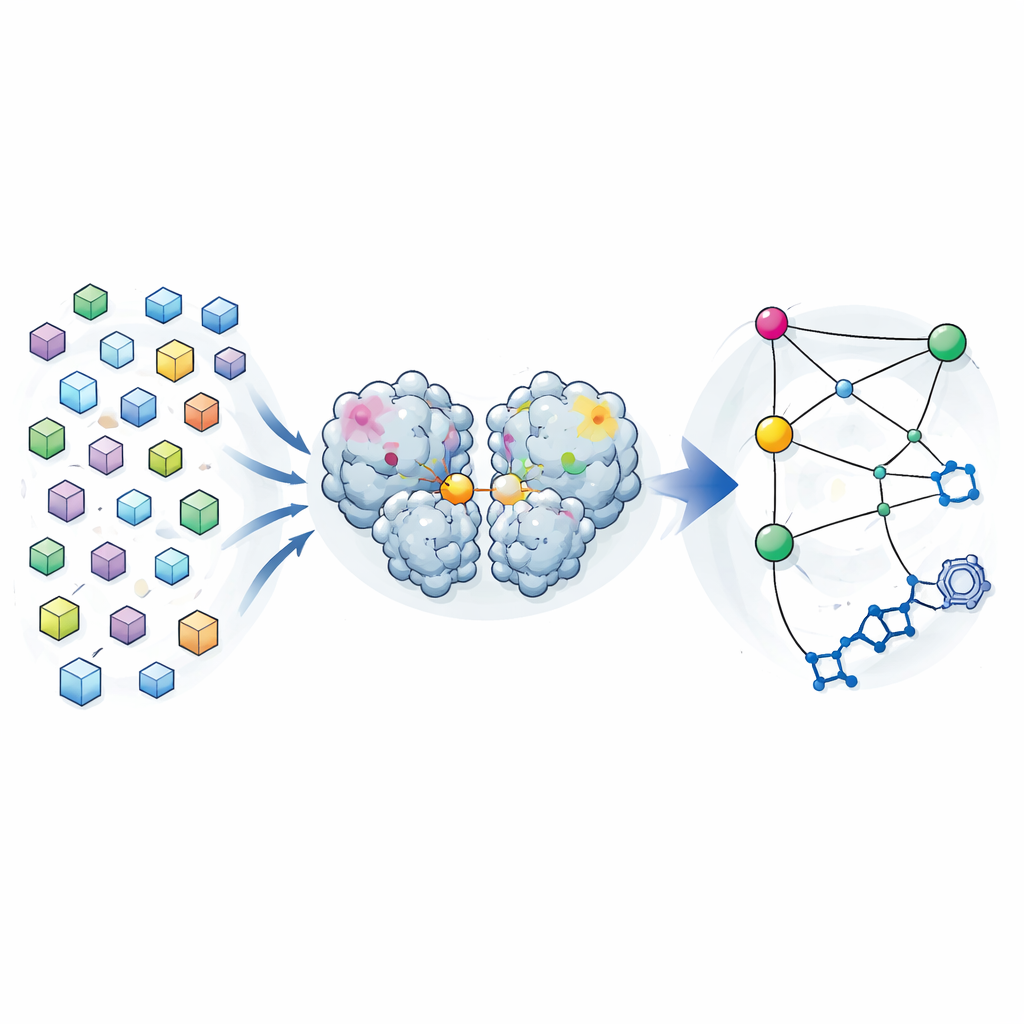
Wiele migawek zamiast jednej struktury
Tradycyjna kryształografia dąży do przedstawienia jednej „najlepszej” struktury białka, uśrednionej po wielu cząsteczkach w krysztale. Autorzy odwrócili tę logikę. Podczas wcześniejszej kampanii poszukiwania leków zebrali dane dyfrakcyjne z niemal 7000 kryształów głównej proteazy SARS-CoV-2. Po starannym odfiltrowaniu kryształów zawierających związane związki i zastosowaniu surowych kryteriów jakości pozostało im 1146 struktur wysokiej rozdzielczości enzymu bez ligandów. Zamiast traktować drobne różnice między tymi strukturami jako szum, przeanalizowali je jako rozkład kształtów, które proteaza może przyjmować, pozostając blisko swojej formy roboczej.
Wykrywanie długodystansowych połączeń wewnątrz enzymu
Porównując zmienność położenia każdego atomu między strukturami, zespół zbudował mapę kowariancji — rodzaj statystycznego odcisku palca pokazującego, które regiony mają tendencję do poruszania się razem. Stwierdzili, że korelacje oparte na kryształach były bardzo podobne do tych przewidywanych przez długie symulacje dynamiki molekularnej proteazy w roztworze. Sugerowało to, że zmienność „zamrożona” w kryształach odzwierciedla prawdziwe ruchy fleksyjne, które enzym eksploruje w wodzie. Skupiając się na korelacjach różnych regionów z centrum aktywnym — kieszenią wykonującą cięcie — odkryli trzy „ogniska” w domenie dymeryzacji proteazy, części pomagającej parować się dwóm kopiom enzymu. Zmiany w tych ogniskach, oddalonych od miejsca aktywnego, okazały się ściśle powiązane ze zmianami w kieszeni katalitycznej.
Testowanie ognisk przez ukierunkowane modyfikacje
Aby sprawdzić, czy statystycznie zidentyfikowane miejsca rzeczywiście kontrolują funkcję, badacze zmienili każde ognisko, zastępując pojedyncze aminokwasy alaniną — standardowym zabiegiem delikatnie zaburzającym lokalną strukturę. Następnie zmierzyli, jak dobrze każdy mutant proteazy tworzy dymery, wiąże fragment podobny do substratu i tnie fluorescencyjny peptyd testowy. Jedna pozycja, N214, zachowywała się jak potwierdzone „ognisko allosteryczne”: mutacja osłabiła tworzenie dymery ponad dziesięciokrotnie, a nawet gdy dymery były obecne, szybkość katalizy spadła około rzędu wielkości. Struktury wysokiej rozdzielczości mutanta wyjaśniły przyczynę: lokalna sieć wiązań wodorowych, która normalnie stabilizuje kluczową pętlę tworzącą „dziurę oksoniową” w miejscu aktywnym, została przerwana, a pętla stała się częściowo nieuporządkowana i niewłaściwie ustawiona do prowadzenia reakcji chemicznej.
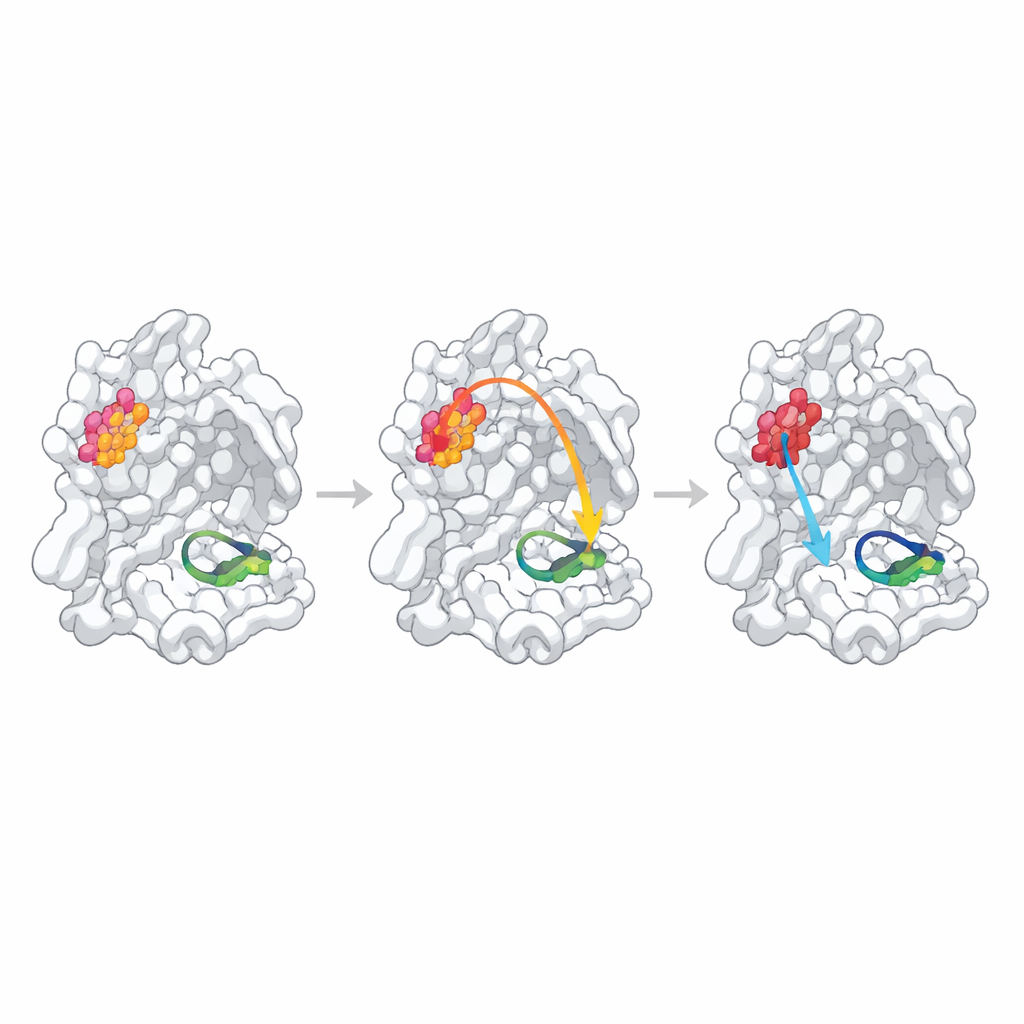
Gdy przewidywane powiązania są słabe lub ukryte
Pozostałe dwa przewidywane ogniska zachowały się inaczej. Zmiana Q256, reszty wystawionej na rozpuszczalnik i pozornie niezaangażowanej w kontakty dymeryczne, spowodowała jedynie umiarkowany spadek aktywności, który można było w dużej mierze wyjaśnić nieznacznie słabszym tworzeniem dymery i nieco gorszym wiązaniem testowego inhibitora. Struktura kryształowa ujawniła zupełnie nowy sposób upakowania, w którym sąsiednie cząsteczki wypełniły szczelinę po mutacji, co utrudnia rozróżnienie prawdziwej wewnętrznej komunikacji od efektów specyficznych dla kryształu. Trzecie miejsce, S284, leży w małym „zameczku” łączącym dwie kopie enzymu. Zgodnie z oczekiwaniami wynikającymi z porównań sekwencyjnych z pokrewnymi koronawirusami, zastąpienie tej reszty miało niemal zerowy wpływ na strukturę i funkcję, wskazując, że ta konkretna zamiana na alaninę była zbyt łagodnym zaburzeniem, by przetestować przewidywaną ścieżkę.
Kryształy jako maleńkie eksperymenty siłowe
Intrygującym ubocznym wynikiem pracy jest to, że wiele różnic strukturalnych między kryształami można przypisać drobnym zmianom w sieci krystalicznej — powtarzalnym układzie białek i rozpuszczalnika. W miarę jak kryształy lekko wysychały podczas manipulacji, ich komórki elementarne kurczyły się lub rozciągały, a te przesunięcia pociągały za punkty kontaktowe między sąsiednimi cząsteczkami proteazy. Te siły rozchodziły się po białku, generując różnorodność strukturalną leżącą u podstaw analizy kowariancji. Innymi słowy, każdy kryształ działał jak mały eksperyment mechaniczny, delikatnie naprężając proteazę w różnych kierunkach i ujawniając, jak się wygina, nie rozkładając się przy tym.
Dlaczego to ma znaczenie dla biologii i projektowania leków
Traktując tysiące struktur krystalicznych jako populację statystyczną, a nie jedną odpowiedź, autorzy mogli wywnioskować sieć allosteryczną łączącą resztę w interfejsie dymeryzacji, N214, z centrum katalitycznym głównej proteazy SARS-CoV-2. Zaburzenie tego miejsca nie tylko zmniejsza liczbę powstających dymery; konkretnie zaburza geometrię pętli aktywnej niezbędnej do wydajnej katalizy. Potwierdza to, że aktywność enzymu jest regulowana przez komunikację na duże odległości w obrębie białka. Szerzej, badanie demonstruje podejście „kryształografii statystycznej”, w którym niekontrolowane, codzienne perturbacje kryształów — takie jak drobne różnice w wysychaniu — stają się zasobem do mapowania ruchów funkcjonalnych. W miarę gromadzenia podobnych zestawów danych dla innych białek ta strategia może pomóc ujawnić nowe miejsca regulacyjne i ukierunkować projektowanie leków, które działają nie tylko przez blokowanie miejsc aktywnych, lecz przez przekształcanie wewnętrznych „konwersacji”, które decydują o działaniu białek.
Cytowanie: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Słowa kluczowe: Główna proteaza SARS-CoV-2, allosteryczność, dynamika białek, kryształografia, poszukiwanie leków