Clear Sky Science · es
La cristalografía estadística revela una red alostérica en Mpro de SARS-CoV-2
Cómo el cambio de forma de las proteínas ayuda a funcionar a una enzima del coronavirus
El virus que causa la COVID-19 depende de una máquina molecular llamada proteasa principal para cortar largas cadenas virales en partes funcionales. Los fármacos ya atacan esta enzima, pero los científicos todavía tienen dificultades para ver cómo sus sutiles movimientos internos controlan cuándo se enciende o apaga. Este estudio usa más de mil instantáneas de la proteasa encerrada en cristales para revelar cómo partes distantes de la molécula se comunican con el centro de trabajo donde ocurre la química—y muestra cómo esta comunicación oculta puede ser perturbada.
Muchas instantáneas en lugar de sólo una
La cristalografía tradicional busca proporcionar una única “mejor” estructura de una proteína, promediada sobre muchas moléculas en un cristal. Los autores invirtieron esta lógica. Durante una campaña previa de búsqueda de fármacos, recopilaron datos de difracción de casi 7000 cristales de la proteasa principal de SARS-CoV-2. Tras filtrar cuidadosamente cualquier cristal que contuviera compuestos unidos y aplicar límites estrictos de calidad, les quedaron 1146 estructuras de alta resolución de la enzima sola. En lugar de tratar las pequeñas diferencias entre estas estructuras como mero ruido, las analizaron como una distribución de formas que la proteasa puede adoptar mientras permanece cerca de su forma funcional.
Encontrar conexiones a larga distancia dentro de la enzima
Al comparar cómo variaba la posición de cada átomo de estructura a estructura, el equipo construyó un mapa de covarianza: una especie de huella estadística que muestra qué regiones tienden a moverse juntas. Encontraron que estas correlaciones basadas en cristales se parecían mucho a las predichas por largas simulaciones de dinámica molecular de la proteasa en solución. Esto sugiere que la variabilidad congelada en los cristales refleja movimientos reales de flexión que la enzima explora en agua. Cuando se centraron en cómo diferentes regiones se correlacionaban con el sitio activo—el bolsillo que realiza los cortes—descubrieron tres “puntos calientes” en el dominio de dimerización de la proteasa, la porción que ayuda a que dos copias de la enzima se emparejen. Los cambios en estos puntos calientes, alejados del sitio activo, parecían estar fuertemente ligados a cambios en el bolsillo catalítico.
Probar los puntos calientes con cambios dirigidos
Para sondear si estos sitios identificados estadísticamente realmente controlan la función, los investigadores alteraron cada punto caliente reemplazando un solo aminoácido por alanina, una manera estándar de modificar ligeramente la estructura local. Luego midieron qué tan bien cada proteasa mutante formaba dímeros, unía un fragmento similar al sustrato y cortaba un péptido de prueba fluorescente. Una posición, N214, se comportó como un “punto caliente alostérico” confirmado: la mutación debilitó la formación de dímeros más de diez veces y, aun cuando había suficientes dímeros presentes, la tasa catalítica cayó aproximadamente un orden de magnitud. Estructuras de alta resolución del mutante mostraron la razón: la red local de enlaces de hidrógeno que normalmente estabiliza un bucle clave que forma el “hueco del oxianión” en el sitio activo se rompió, y ese bucle se volvió parcialmente desordenado y desalineado para la química.
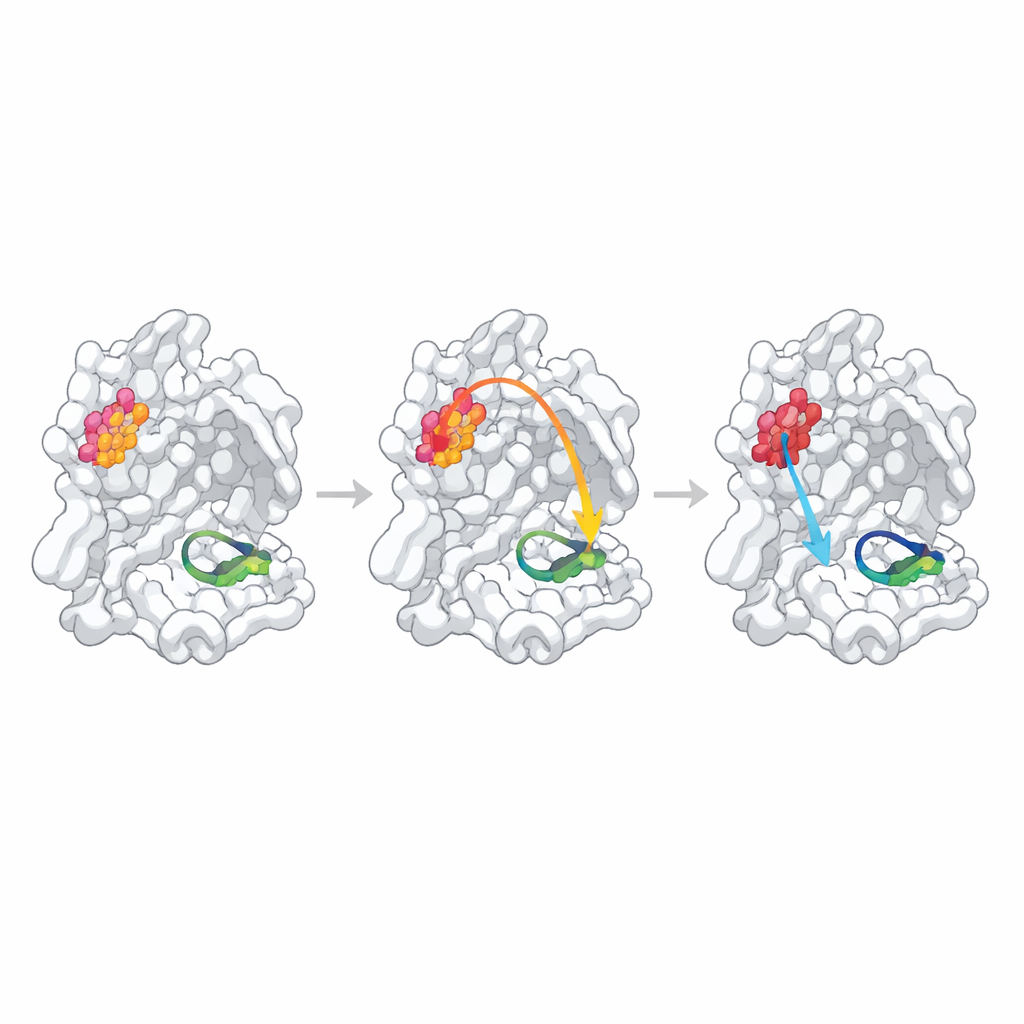
Cuando los enlaces predichos son débiles u ocultos
Los otros dos puntos calientes predichos se comportaron de manera diferente. Cambiar Q256, un residuo expuesto al solvente y no evidentemente involucrado en contactos de dimerización, produjo sólo una disminución modesta de la actividad que podía explicarse en gran medida por una formación de dímeros ligeramente más débil y una unión algo menor del inhibidor de prueba. Su estructura cristalina reveló un arreglo de empaquetamiento completamente nuevo donde moléculas vecinas llenaron la cavidad dejada por la mutación, lo que dificultó distinguir la verdadera comunicación interna de efectos específicos del cristal. El tercer sitio, S284, se sitúa en una pequeña “cremallera” que conecta las dos copias de la enzima. Como era de esperar por comparaciones de secuencia con coronavirus relacionados, reemplazar este residuo tuvo casi ningún efecto en la estructura o la función, indicando que este cambio por alanina en particular fue una perturbación demasiado suave para probar la vía predicha.
Cristales como pequeños experimentos de fuerza
Un resultado lateral intrigante del trabajo es que muchas de las diferencias estructurales entre cristales pudieron rastrearse a pequeños cambios en la red cristalina: la disposición repetitiva de proteínas y solvente. A medida que los cristales se secaron en grados ligeramente distintos durante la manipulación, sus celdas unitarias se contrajeron o estiraron, y estos cambios tiraron de los puntos de contacto entre moléculas vecinas de proteasa. Esas fuerzas se propagaron a través de la proteína, generando la diversidad estructural que sustentó el análisis de covarianza. En otras palabras, cada cristal actuó como un pequeño experimento mecánico, tensando suavemente la proteasa en distintas direcciones y revelando cómo se flexibiliza sin desmoronarse.
Por qué esto importa para la biología y el diseño de fármacos
Al tratar miles de estructuras cristalinas como una población estadística en lugar de una sola respuesta, los autores pudieron inferir una red alostérica que enlaza un residuo de la interfaz de dimerización, N214, con el centro catalítico de la proteasa principal de SARS-CoV-2. Perturbar este sitio no solo reduce cuántos dímeros se forman; desordena específicamente la geometría del bucle activo necesaria para una catálisis eficiente. Esto confirma que la actividad de la enzima está gobernada por comunicación a larga distancia a través de la proteína. Más ampliamente, el estudio demuestra un enfoque de “cristalografía estadística” en el que perturbaciones cotidianas no controladas a los cristales—como pequeñas diferencias en el secado—se convierten en un recurso para cartografiar movimentos funcionales. A medida que se recojan conjuntos de datos similares para otras proteínas, esta estrategia podría ayudar a descubrir nuevos sitios regulatorios y guiar el diseño de fármacos que actúen no solo bloqueando sitios activos, sino reconfigurando las conversaciones internas que hacen funcionar a las proteínas.
Cita: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Palabras clave: proteasa principal de SARS-CoV-2, alosteria, dinámica de proteínas, cristalografía, descubrimiento de fármacos