Clear Sky Science · nl
Statistische kristallografie onthult een allosterisch netwerk in SARS-CoV-2 Mpro
Hoe het van vorm veranderen van eiwitten een coronavirus-enzym helpt werken
Het virus dat COVID-19 veroorzaakt, is afhankelijk van een moleculaire machine die het hoofdprotease wordt genoemd om lange virale ketens in werkende onderdelen te knippen. Geneesmiddelen richten zich al op dit enzym, maar wetenschappers hebben nog moeite om te begrijpen hoe subtiele interne bewegingen bepalen wanneer het aan- of uitgezet wordt. Deze studie gebruikt meer dan duizend momentopnamen van het protease vastgelegd in kristallen om te laten zien hoe verre delen van het molecuul communiceren met het werkcentrum waar de chemie plaatsvindt — en toont aan hoe deze verborgen communicatie verstoord kan worden.
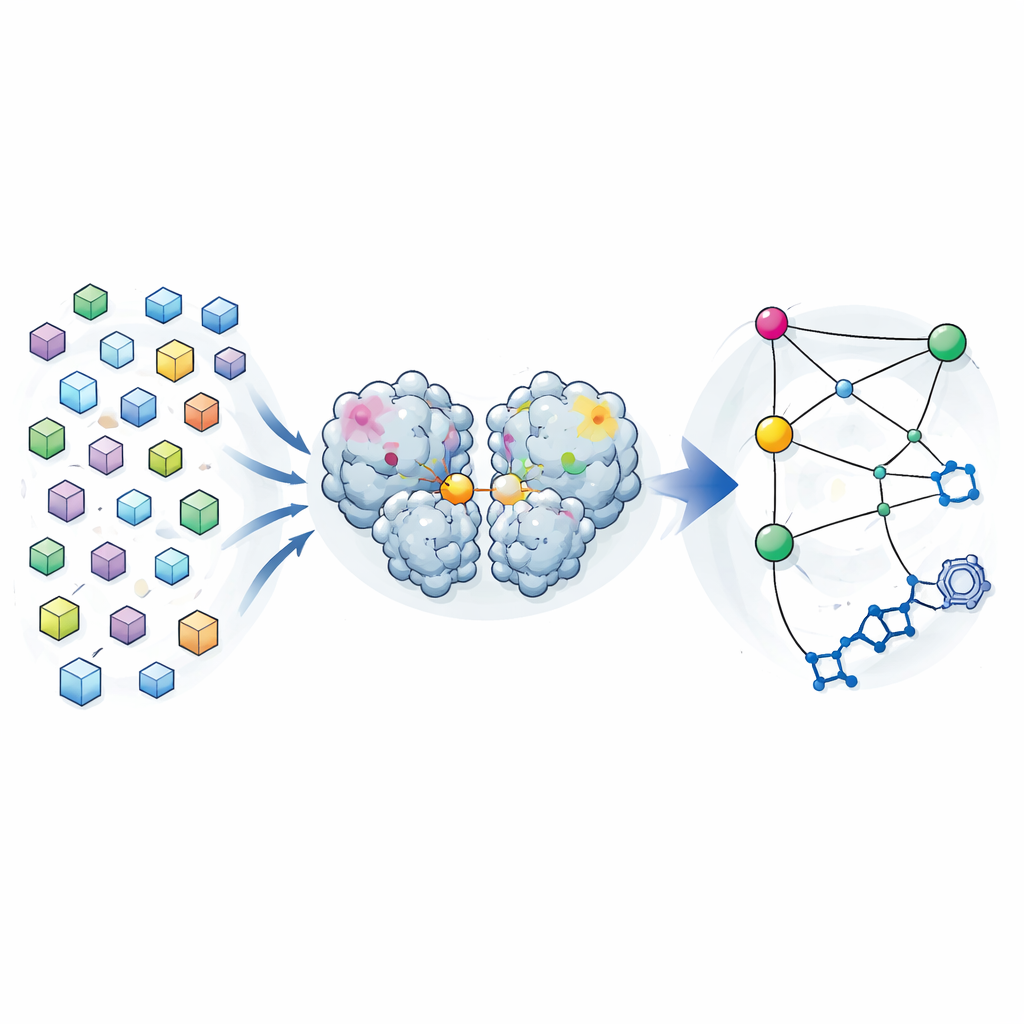
Vele momentopnamen in plaats van slechts één
Traditionele kristallografie streeft ernaar één “beste” structuur van een eiwit te leveren, gemiddeld over veel moleculen in een kristal. De auteurs draaiden deze logica om. Tijdens een eerdere zoektocht naar geneesmiddelen hadden ze diffractiegegevens verzameld van bijna 7000 kristallen van het SARS-CoV-2 hoofdprotease. Na zorgvuldig alle kristallen die gebonden verbindingen bevatten eruit te filteren en strikte kwaliteitslimieten toe te passen, bleven ze over met 1146 hogeresolutiestructuren van het enzym alleen. In plaats van kleine verschillen tussen deze structuren als ruis te behandelen, analyseerden ze ze als een verdeling van vormen die het protease kan aannemen terwijl het dicht bij zijn werkende vorm blijft.
Het vinden van langafstandsverbindingen binnen het enzym
Door te vergelijken hoe de positie van elk atoom van structuur tot structuur varieerde, bouwde het team een covariantiemap — een soort statistische vingerafdruk die toont welke regio’s de neiging hebben samen te bewegen. Ze vonden dat deze op kristal gebaseerde correlaties sterk leken op die voorspeld door lange moleculaire-dynamicasimulaties van het protease in oplossing. Dit suggereerde dat de variabiliteit die in de kristallen bevroren zit, echte buigbewegingen weergeeft die het enzym in water verkent. Toen ze zich richtten op hoe verschillende regio’s correleerden met het actieve centrum — het zakje dat het knippen doet — ontdekten ze drie “hotspots” in het dimerisatiedomein van het protease, het deel dat helpt twee kopieën van het enzym te laten samenkomen. Veranderingen op deze hotspots, ver weg van het actieve centrum, leken sterk gekoppeld aan veranderingen in het katalytische zakje.
De hotspots testen met gerichte veranderingen
Om te onderzoeken of deze statistisch geïdentificeerde plaatsen daadwerkelijk de functie controleren, wijzigden de onderzoekers elke hotspot door één aminozuur te vervangen door alanine, een gangbare manier om de lokale structuur licht te beïnvloeden. Ze maten vervolgens hoe goed elk gemuteerd protease dimers vormde, een substraatachtig fragment bond, en een fluorescent testpeptide knipte. Één positie, N214, gedroeg zich als een bevestigd “allosterisch hotspot”: de mutatie verzwakte dimervorming meer dan tienvoudig en, zelfs wanneer er genoeg dimers aanwezig waren, daalde de katalytische snelheid met ongeveer een orde van grootte. Hogeresolutiestructuren van de mutant toonden waarom: het lokale waterstofbruggennetwerk dat normaal een cruciale lus stabiliseert die het “oxyaniongaten” in het actieve centrum vormt, was verbroken, en deze lus werd gedeeltelijk gedesordend en verkeerd uitgelijnd voor chemie.
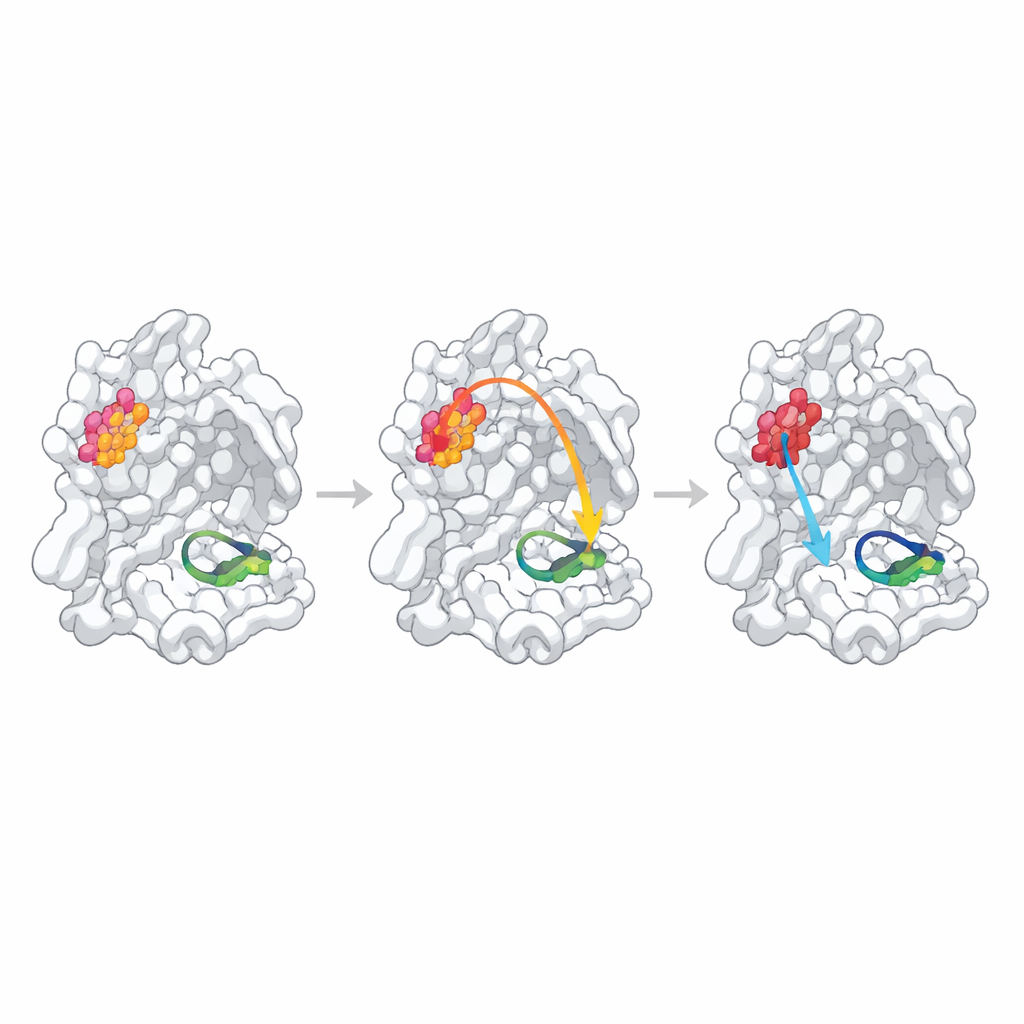
Wanneer voorspelde verbindingen zwak of verborgen zijn
De andere twee voorspelde hotspots gedroegen zich anders. Het veranderen van Q256, een residu dat aan het oplosmiddel blootgesteld is en niet duidelijk betrokken bij dimercontacten, veroorzaakte slechts een bescheiden afname in activiteit die grotendeels verklaard kon worden door iets zwakkere dimervorming en enigszins slechtere binding van de testremmer. De kristalstructuur toonde een volledig nieuwe pakkingindeling waarin naburige moleculen de holte opvulden die door de mutatie was achtergelaten, waardoor het moeilijk werd om echte interne communicatie te onderscheiden van kristalspecifieke effecten. De derde plaats, S284, ligt in een kleine “rits” die de twee enzymkopieën met elkaar verbindt. Zoals verwacht op basis van sequentievergelijkingen met verwante coronavirussen, had het vervangen van dit residu vrijwel geen effect op structuur of functie, wat aangeeft dat deze specifieke alanineverandering te mild was om het voorspelde pad te testen.
Kristallen als kleine kracht-experimenten
Een intrigerend bijproduct van het werk is dat veel van de structurele verschillen tussen kristallen terug te voeren waren op kleine veranderingen in het kristalrooster — de herhalende rangschikking van eiwitten en oplosmiddel. Terwijl kristallen tijdens hantering in licht verschillende mate uitdroogden, krimpten of strekten hun eenheidscellen, en deze verschuivingen trokken aan contactpunten tussen naburige proteasemoleculen. Die krachten rimpelden door het eiwit en genereerden de structurele diversiteit die de covariantieanalyse ondersteunde. Met andere woorden: elk kristal fungeerde als een klein mechanisch experiment, dat het protease zachtjes langs verschillende richtingen belaste en toonde hoe het buigt zonder uit elkaar te vallen.
Waarom dit belangrijk is voor biologie en geneesmiddelenontwerp
Door duizenden kristalstructuren te behandelen als een statistische populatie in plaats van als één antwoord, konden de auteurs een allosterisch netwerk afleiden dat een residu in het dimerinterfaces, N214, koppelt aan het katalytische centrum van het SARS-CoV-2 hoofdprotease. Het verstoren van deze plaats vermindert niet alleen het aantal gevormde dimers; het verstoort specifiek de geometrie van de actieve lus die nodig is voor efficiënte katalyse. Dit bevestigt dat de activiteit van het enzym wordt geregeld door langafstandscommunicatie door het eiwit. In bredere zin toont de studie een “statistische kristallografie”-benadering aan waarbij ongecontroleerde, alledaagse verstoringen van kristallen — zoals kleine verschillen in uitdroging — een bron worden om functionele bewegingen in kaart te brengen. Naarmate soortgelijke datasets voor andere eiwitten worden verzameld, kan deze strategie helpen nieuwe regulerende plaatsen te ontdekken en het ontwerp van geneesmiddelen sturen die niet alleen werken door actieve centra te blokkeren, maar door de interne gesprekken die eiwitten laten werken, te herbedraden.
Bronvermelding: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Trefwoorden: SARS-CoV-2 hoofdprotease, allosterie, proteïnedynamica, kristallografie, geneesmiddelenontdekking