Clear Sky Science · en
Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro
How Protein Shape-Shifting Helps a Coronavirus Enzyme Work
The virus that causes COVID-19 depends on a molecular machine called the main protease to cut long viral chains into working parts. Drugs already target this enzyme, but scientists still struggle to see how its subtle internal motions control when it switches on or off. This study uses more than a thousand snapshots of the protease locked in crystals to reveal how distant parts of the molecule talk to the business center where chemistry happens—and shows how this hidden communication can be disrupted.
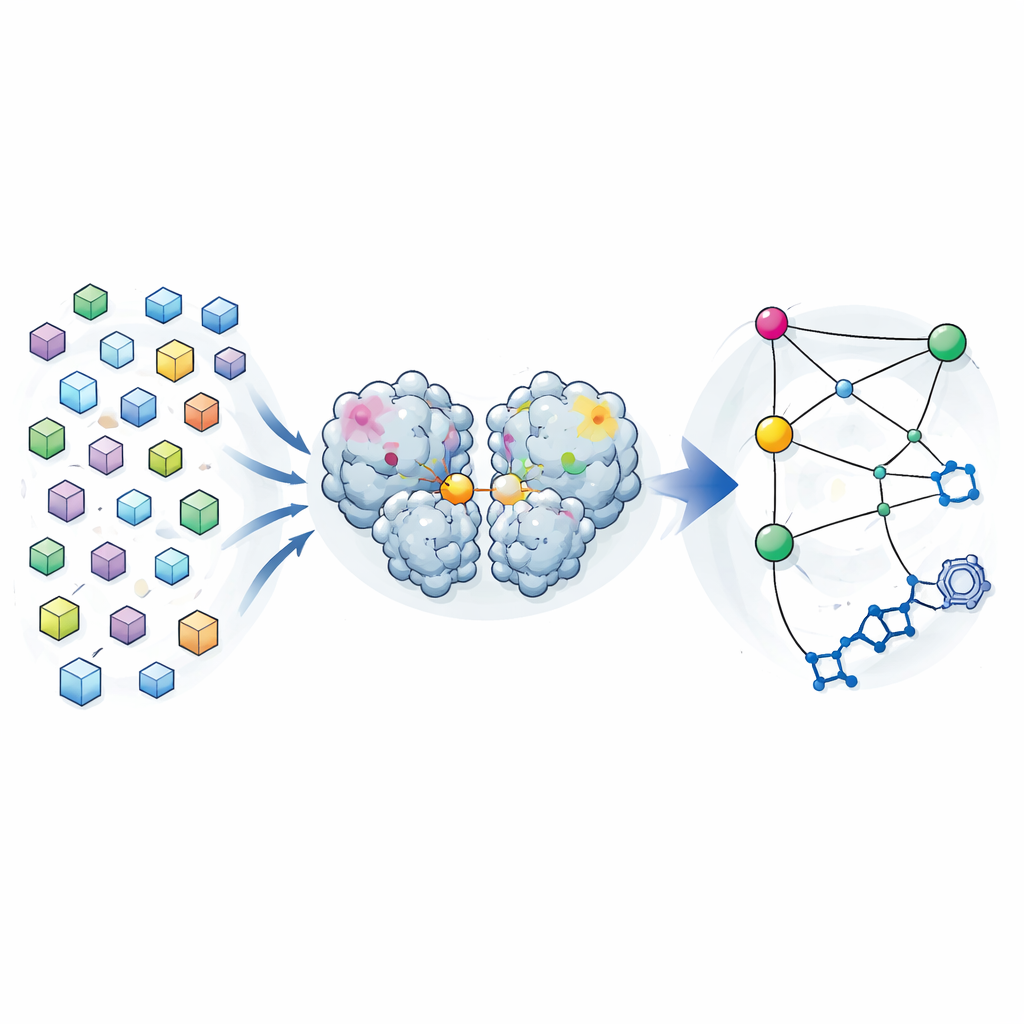
Many Snapshots Instead of Just One
Traditional crystallography aims to provide a single “best” structure of a protein, averaged over many molecules in a crystal. The authors turned this logic on its head. During an earlier drug-search campaign, they had collected diffraction data from nearly 7000 crystals of the SARS-CoV-2 main protease. After carefully filtering out any crystals that contained bound compounds and enforcing strict quality limits, they were left with 1146 high-resolution structures of the enzyme alone. Rather than treating small differences between these structures as mere noise, they analyzed them as a distribution of shapes that the protease can adopt while remaining close to its working form.
Finding Long-Distance Connections Inside the Enzyme
By comparing how each atom’s position varied from structure to structure, the team built a covariance map—a kind of statistical fingerprint showing which regions tend to move together. They found that these crystal-based correlations closely resembled those predicted by long molecular dynamics simulations of the protease in solution. This suggested that the variability frozen into the crystals reflects real flexing motions the enzyme explores in water. When they focused on how different regions correlated with the active site—the pocket that does the cutting—they uncovered three “hotspots” in the protease’s dimerization domain, the portion that helps two copies of the enzyme pair up. Changes at these hotspots, far from the active site, appeared to be tightly linked to changes in the catalytic pocket.
Testing the Hotspots with Targeted Changes
To probe whether these statistically identified sites truly control function, the researchers altered each hotspot by replacing a single amino acid with alanine, a standard way to nudge local structure. They then measured how well each mutant protease formed dimers, bound a substrate-like fragment, and cut a fluorescent test peptide. One position, N214, behaved as a confirmed “allosteric hotspot”: the mutation weakened dimer formation more than tenfold and, even when enough dimers were present, the catalytic rate dropped by about an order of magnitude. High-resolution structures of the mutant showed why: the local hydrogen-bond network that normally stabilizes a crucial loop forming the “oxyanion hole” in the active site was broken, and this loop became partially disordered and misaligned for chemistry.
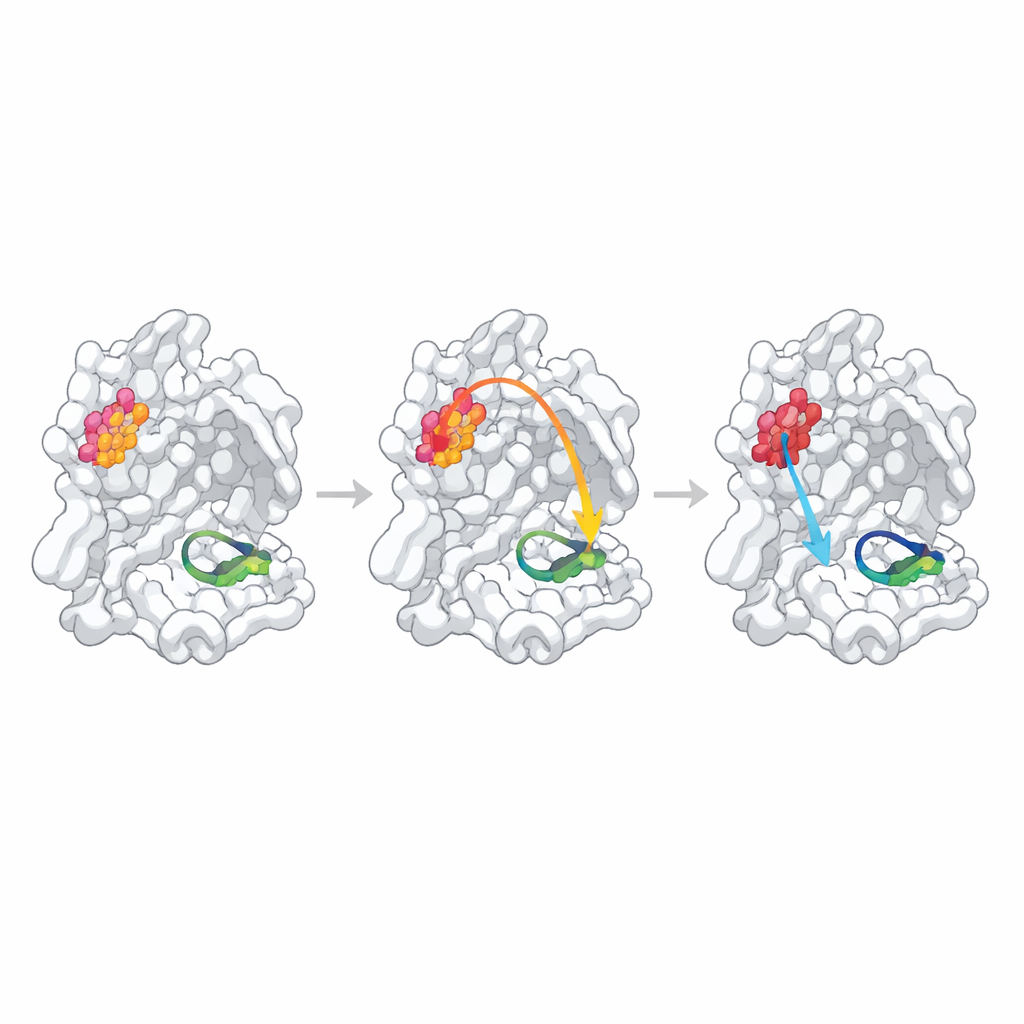
When Predicted Links Are Weak or Hidden
The other two predicted hotspots behaved differently. Changing Q256, a residue exposed to solvent and not obviously involved in dimer contacts, produced only a modest decrease in activity that could be explained largely by slightly weaker dimer formation and somewhat poorer binding of the test inhibitor. Its crystal structure revealed a completely new packing arrangement where neighboring molecules filled the cavity left by the mutation, making it difficult to distinguish true internal communication from crystal-specific effects. The third site, S284, lies in a small “zipper” that bridges the two enzyme copies. As expected from sequence comparisons with related coronaviruses, replacing this residue had almost no effect on structure or function, indicating that this particular alanine change was too gentle a perturbation to test the predicted pathway.
Crystals as Tiny Force Experiments
An intriguing side result of the work is that many of the structural differences between crystals could be traced to tiny changes in the crystal lattice—the repeating arrangement of proteins and solvent. As crystals dried to slightly different degrees during handling, their unit cells shrank or stretched, and these shifts tugged on contact points between neighboring protease molecules. Those forces rippled through the protein, generating the structural diversity that underpinned the covariance analysis. In other words, each crystal acted as a small mechanical experiment, gently straining the protease along different directions and revealing how it flexes without falling apart.
Why This Matters for Biology and Drug Design
By treating thousands of crystal structures as a statistical population rather than a single answer, the authors could infer an allosteric network that links a dimer interface residue, N214, to the catalytic center of the SARS-CoV-2 main protease. Disrupting this site does not simply reduce how many dimers form; it specifically scrambles the geometry of the active loop required for efficient catalysis. This confirms that the enzyme’s activity is governed by long-range communication across the protein. More broadly, the study demonstrates a “statistical crystallography” approach in which uncontrolled, everyday perturbations to crystals—such as slight differences in drying—become a resource for mapping functional motions. As similar datasets are gathered for other proteins, this strategy could help uncover new regulatory sites and guide the design of drugs that work not just by plugging active sites but by rewiring the internal conversations that make proteins tick.
Citation: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Keywords: SARS-CoV-2 main protease, allostery, protein dynamics, crystallography, drug discovery