Clear Sky Science · fr
La cristallographie statistique révèle un réseau allostérique dans la Mpro du SARS-CoV-2
Comment le changement de forme des protéines aide une enzyme de coronavirus à fonctionner
Le virus responsable de la COVID-19 dépend d’une machinerie moléculaire appelée protéase principale pour couper de longues chaînes virales en éléments fonctionnels. Des médicaments ciblent déjà cette enzyme, mais les chercheurs peinent encore à voir comment ses mouvements internes subtils contrôlent quand elle s’active ou se désactive. Cette étude utilise plus d’un millier d’instantanés de la protéase enfermée dans des cristaux pour révéler comment des parties éloignées de la molécule communiquent avec le centre d’activité chimique — et montre comment cette communication cachée peut être perturbée.
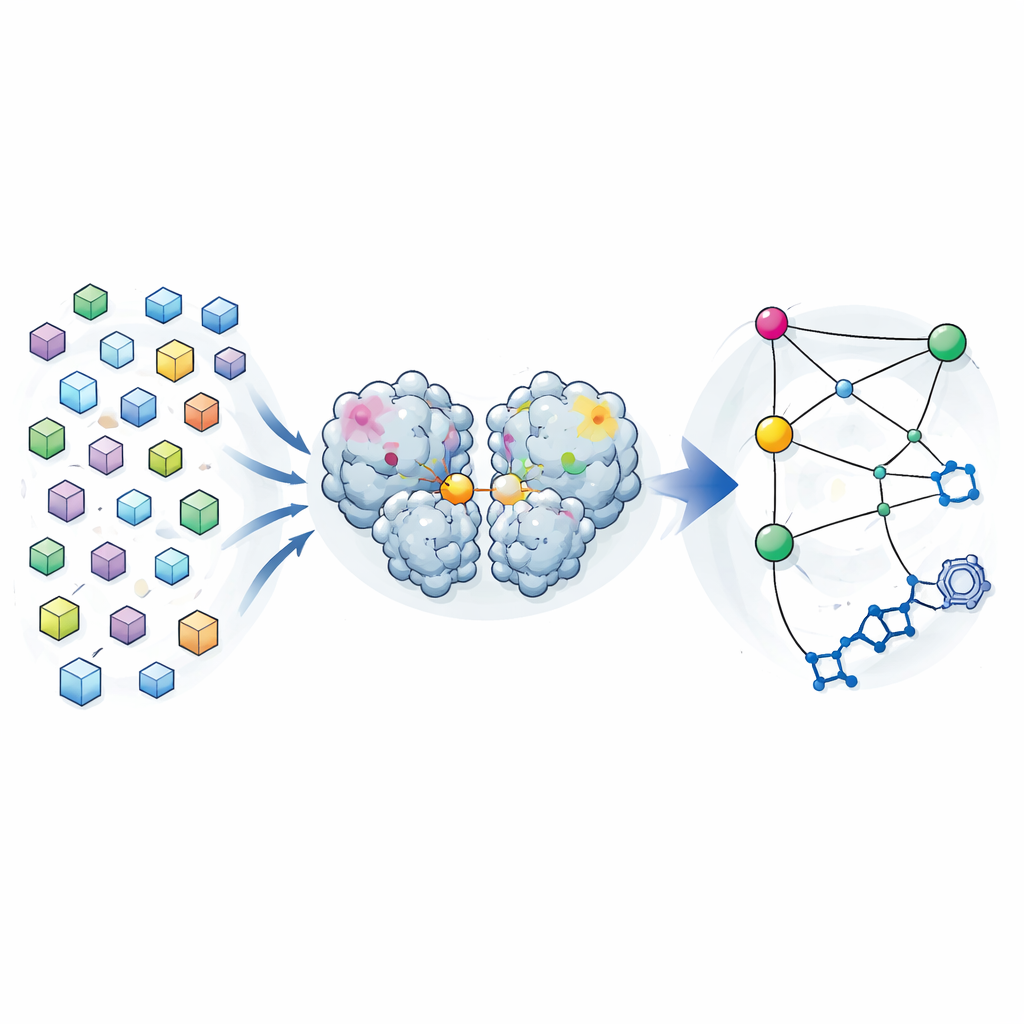
Beaucoup d’instantanés au lieu d’un seul
La cristallographie traditionnelle vise à fournir une « meilleure » structure unique d’une protéine, moyennée sur de nombreuses molécules dans un cristal. Les auteurs ont renversé cette logique. Lors d’une campagne précédente de recherche de médicaments, ils avaient collecté des données de diffraction provenant de près de 7000 cristaux de la protéase principale du SARS-CoV-2. Après avoir soigneusement filtré tous les cristaux contenant des composés liés et appliqué des limites de qualité strictes, il leur restait 1146 structures haute résolution de l’enzyme seule. Plutôt que de traiter les petites différences entre ces structures comme un simple bruit, ils les ont analysées comme une distribution de formes que la protéase peut adopter tout en restant proche de sa forme active.
Identifier des connexions à longue distance à l’intérieur de l’enzyme
En comparant la manière dont la position de chaque atome variait d’une structure à l’autre, l’équipe a construit une carte de covariance — une sorte d’empreinte statistique montrant quelles régions ont tendance à bouger ensemble. Ils ont constaté que ces corrélations issues des cristaux ressemblaient étroitement à celles prédites par de longues simulations de dynamique moléculaire de la protéase en solution. Cela suggérait que la variabilité figée dans les cristaux reflète de réels mouvements de flexion que l’enzyme explore dans l’eau. Lorsqu’ils se sont concentrés sur la corrélation entre différentes régions et le site actif — la poche qui réalise la coupure — ils ont mis au jour trois « points chauds » dans le domaine de dimérisation de la protéase, la portion qui aide deux copies de l’enzyme à se jumeler. Des changements à ces points chauds, éloignés du site actif, semblaient étroitement liés à des modifications de la poche catalytique.
Tester les points chauds avec des changements ciblés
Pour vérifier si ces sites identifiés statistiquement contrôlaient réellement la fonction, les chercheurs ont modifié chaque point chaud en remplaçant un seul acide aminé par de l’alanine, une méthode standard pour provoquer un léger déplacement local de la structure. Ils ont ensuite mesuré la capacité de chaque protéase mutante à former des dimères, à lier un fragment ressemblant au substrat et à couper un peptide test fluorescent. Une position, N214, s’est révélée être un « point chaud allostérique » confirmé : la mutation a affaibli la formation de dimères de plus d’un facteur dix et, même lorsque suffisamment de dimères étaient présents, la vitesse catalytique a chuté d’environ un ordre de grandeur. Des structures haute résolution du mutant ont expliqué pourquoi : le réseau local de liaisons hydrogène qui stabilise normalement une boucle cruciale formant le « trou oxyanionique » dans le site actif était rompu, et cette boucle est devenue partiellement désordonnée et mal alignée pour la réaction chimique.
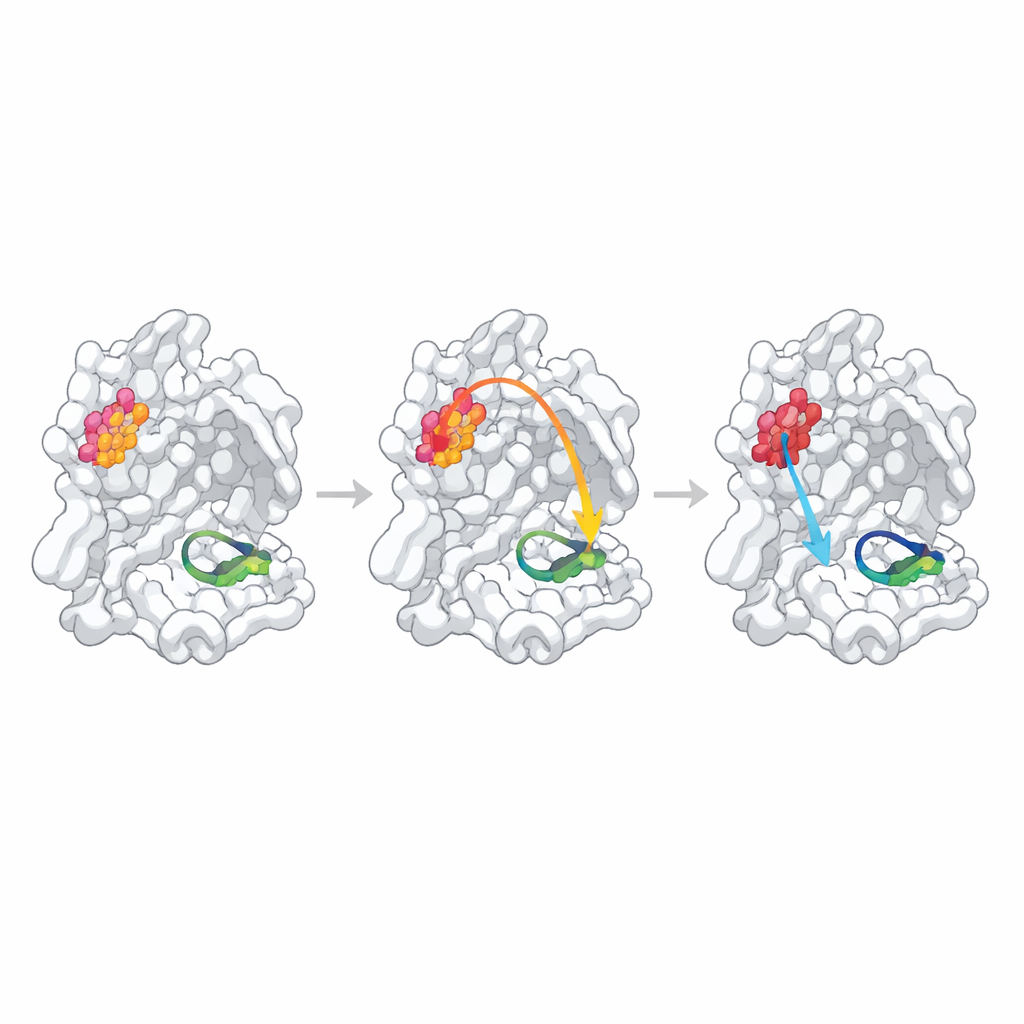
Quand les liens prédits sont faibles ou masqués
Les deux autres points chauds prédits se sont comportés différemment. La modification de Q256, un résidu exposé au solvant et pas visiblement impliqué dans les contacts de dimérisation, n’a entraîné qu’une diminution modeste de l’activité, expliqueable en grande partie par une formation de dimères légèrement plus faible et une affinité un peu moindre pour l’inhibiteur test. Sa structure cristalline a révélé un empilement complètement nouveau où des molécules voisines comblaient la cavité laissée par la mutation, rendant difficile la distinction entre une véritable communication interne et des effets spécifiques au cristal. Le troisième site, S284, se situe dans une petite « fermeture éclair » qui relie les deux copies de l’enzyme. Comme attendu d’après les comparaisons de séquences avec des coronavirus apparentés, remplacer ce résidu n’a presque pas affecté la structure ou la fonction, indiquant que ce changement en alanine était trop discret pour tester la voie prédite.
Les cristaux comme de petites expériences mécaniques
Un résultat secondaire intéressant du travail est que bon nombre des différences structurelles entre cristaux pouvaient être attribuées à de minuscules changements dans le réseau cristallin — l’agencement répétitif des protéines et du solvant. À mesure que les cristaux séchaient à des degrés légèrement différents pendant la manipulation, leurs cellules unitaires se contractaient ou s’étiraient, et ces décalages tiraient sur les points de contact entre molécules de protéase voisines. Ces forces se sont propagées à travers la protéine, générant la diversité structurelle qui a été la base de l’analyse de covariance. En d’autres termes, chaque cristal agissait comme une petite expérience mécanique, soumettant délicatement la protéase à des contraintes dans différentes directions et révélant comment elle fléchit sans se désassembler.
Pourquoi cela compte pour la biologie et la conception de médicaments
En traitant des milliers de structures cristallines comme une population statistique plutôt qu’une réponse unique, les auteurs ont pu inférer un réseau allostérique reliant un résidu de l’interface de dimère, N214, au centre catalytique de la protéase principale du SARS-CoV-2. La perturbation de ce site ne réduit pas simplement le nombre de dimères ; elle perturbe spécifiquement la géométrie de la boucle active nécessaire à une catalyse efficace. Cela confirme que l’activité de l’enzyme est régie par une communication à longue portée à travers la protéine. Plus largement, l’étude démontre une approche de « cristallographie statistique » dans laquelle des perturbations quotidiennes et incontrôlées des cristaux — telles que de légères différences de dessiccation — deviennent une ressource pour cartographier les mouvements fonctionnels. À mesure que des jeux de données similaires seront rassemblés pour d’autres protéines, cette stratégie pourrait aider à découvrir de nouveaux sites régulateurs et guider la conception de médicaments qui agissent non seulement en bouchant les sites actifs mais en reconfigurant les conversations internes qui font fonctionner les protéines.
Citation: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Mots-clés: protéase principale du SARS-CoV-2, allostérie, dynamique des protéines, cristallographie, découverte de médicaments