Clear Sky Science · pt
Cristalografia estatística revela uma rede alostérica na Mpro do SARS-CoV-2
Como a mudança de forma das proteínas ajuda uma enzima do coronavírus a funcionar
O vírus que causa a COVID-19 depende de uma máquina molecular chamada protease principal para cortar longas cadeias virais em partes funcionais. Medicamentos já miram essa enzima, mas os cientistas ainda têm dificuldade em enxergar como seus sutis movimentos internos controlam quando ela liga ou desliga. Este estudo usa mais de mil instantâneos da protease presa em cristais para revelar como partes distantes da molécula se comunicam com o centro de atividade onde a química acontece — e mostra como essa comunicação oculta pode ser perturbada.
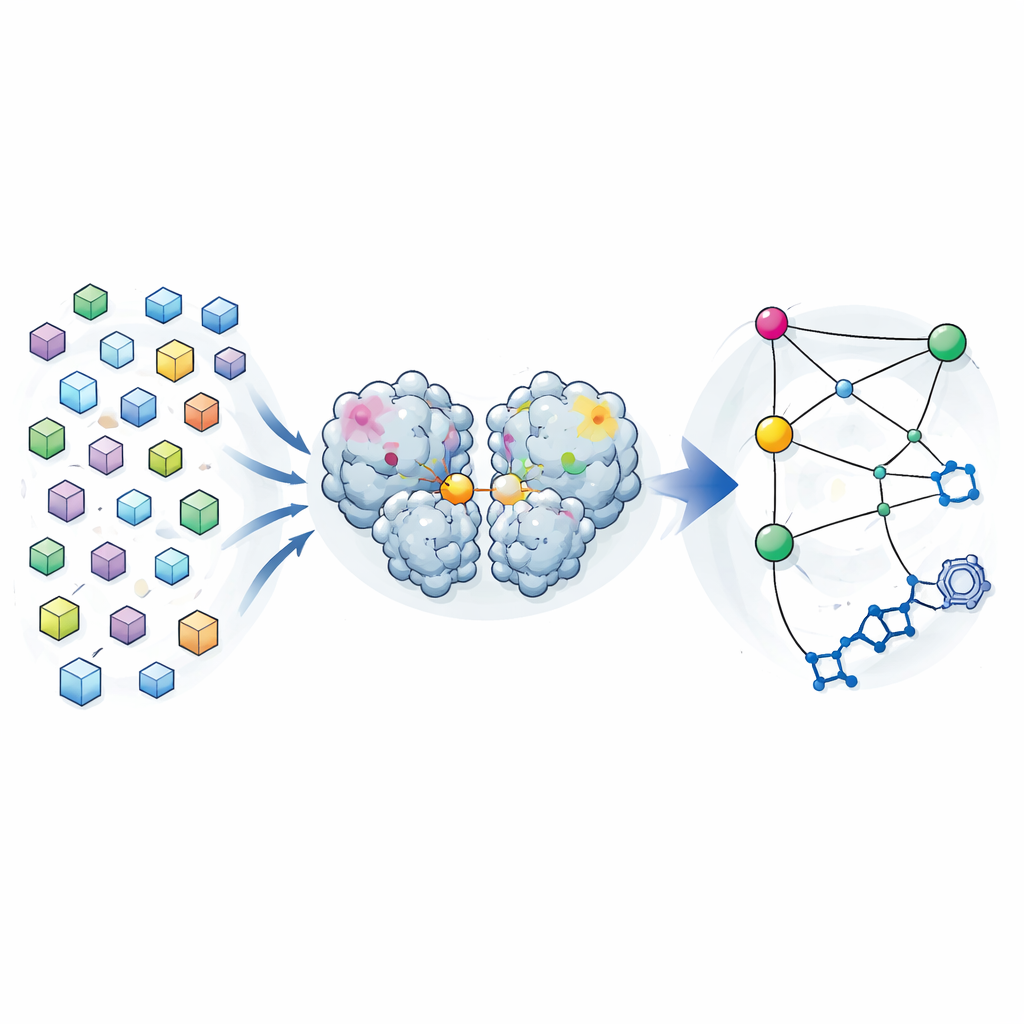
Muitos instantâneos em vez de apenas um
A cristalografia tradicional busca fornecer uma única “melhor” estrutura de uma proteína, média de muitas moléculas no cristal. Os autores inverteram essa lógica. Durante uma campanha anterior de busca de fármacos, eles coletaram dados de difração de quase 7.000 cristais da protease principal do SARS-CoV-2. Após filtrar cuidadosamente quaisquer cristais que continham compostos ligados e impor limites rigorosos de qualidade, restaram 1.146 estruturas de alta resolução da enzima isolada. Em vez de tratar pequenas diferenças entre essas estruturas como mero ruído, eles as analisaram como uma distribuição de formas que a protease pode adotar enquanto permanece próxima de sua forma funcional.
Encontrando conexões de longa distância dentro da enzima
Comparando como a posição de cada átomo variava de estrutura para estrutura, a equipe construiu um mapa de covariância — uma espécie de impressão digital estatística que mostra quais regiões tendem a se mover em conjunto. Eles descobriram que essas correlações baseadas em cristal se assemelhavam fortemente às previstas por longas simulações de dinâmica molecular da protease em solução. Isso sugere que a variabilidade congelada nos cristais reflete movimentos reais de flexão que a enzima explora em água. Quando se concentraram em como diferentes regiões se correlacionavam com o sítio ativo — a bolsa que realiza o corte — desvelaram três “pontos quentes” no domínio de dimerização da protease, a porção que ajuda duas cópias da enzima a se emparelhar. Mudanças nesses pontos quentes, longe do sítio ativo, pareciam estar intimamente ligadas às alterações no bolso catalítico.
Testando os pontos quentes com alterações direcionadas
Para sondar se esses sítios identificados estatisticamente controlam realmente a função, os pesquisadores alteraram cada ponto quente substituindo um único aminoácido por alanina, uma forma padrão de perturbar levemente a estrutura local. Em seguida mediram quão bem cada protease mutante formava dímeros, ligava um fragmento semelhante a substrato e cortava um peptídeo teste fluorescente. Uma posição, N214, comportou-se como um “ponto quente alostérico” confirmado: a mutação enfraqueceu a formação de dímeros em mais de dez vezes e, mesmo quando dímeros suficientes estavam presentes, a taxa catalítica caiu cerca de uma ordem de magnitude. Estruturas de alta resolução do mutante mostraram por quê: a rede local de ligações de hidrogênio que normalmente estabiliza um laço crucial formando o “buraco oxianíon” no sítio ativo foi rompida, e esse laço tornou-se parcialmente desordenado e desalinhado para a química.
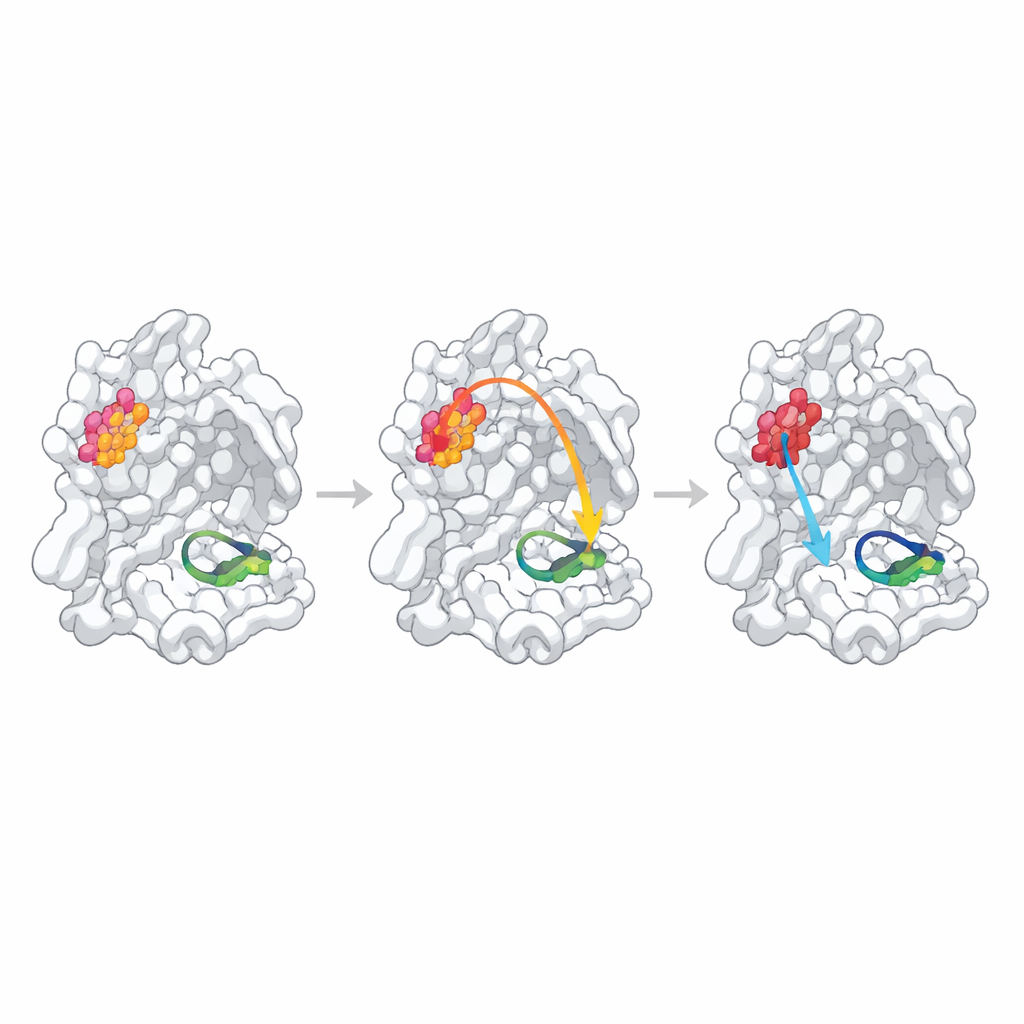
Quando as ligações previstas são fracas ou ocultas
Os outros dois pontos quentes previstos comportaram-se de modo diferente. A mudança em Q256, um resíduo exposto ao solvente e não obviamente envolvido em contatos de dimerização, produziu apenas uma diminuição modesta na atividade que pôde ser explicada em grande parte por formação levemente mais fraca de dímeros e ligação um pouco pior do inibidor de teste. Sua estrutura cristalina revelou um arranjo de empacotamento completamente novo em que moléculas vizinhas preencheram a cavidade deixada pela mutação, tornando difícil distinguir comunicação interna verdadeira de efeitos específicos do cristal. O terceiro sítio, S284, situa-se em um pequeno “zíper” que liga as duas cópias da enzima. Como esperado a partir de comparações de sequência com coronavírus relacionados, substituir esse resíduo teve quase nenhum efeito na estrutura ou função, indicando que essa mudança para alanina foi uma perturbação suave demais para testar a via prevista.
Cristais como pequenos experimentos de força
Um resultado lateral intrigante do trabalho é que muitas das diferenças estruturais entre cristais puderam ser rastreadas até pequenas mudanças na rede cristalina — o arranjo repetitivo de proteínas e solvente. Conforme os cristais secavam em graus ligeiramente diferentes durante o manuseio, suas células unitárias encolhiam ou esticavam, e essas variações puxavam pontos de contato entre moléculas vizinhas da protease. Essas forças propagavam-se pela proteína, gerando a diversidade estrutural que sustentou a análise de covariância. Em outras palavras, cada cristal atuou como um pequeno experimento mecânico, tensionando levemente a protease em diferentes direções e revelando como ela flexiona sem se desfazer.
Por que isso importa para a biologia e o design de fármacos
Ao tratar milhares de estruturas cristalinas como uma população estatística em vez de uma única resposta, os autores puderam inferir uma rede alostérica que liga um resíduo na interface de dímero, N214, ao centro catalítico da protease principal do SARS-CoV-2. Disparar esse sítio não reduz simplesmente o número de dímeros formados; ele embaralha especificamente a geometria do laço ativo necessária para uma catálise eficiente. Isso confirma que a atividade da enzima é governada por comunicação de longo alcance através da proteína. De modo mais amplo, o estudo demonstra uma abordagem de “cristalografia estatística” na qual perturbações cotidianas e não controladas dos cristais — como pequenas diferenças na secagem — tornam-se um recurso para mapear movimentos funcionais. À medida que conjuntos de dados semelhantes forem reunidos para outras proteínas, essa estratégia pode ajudar a descobrir novos sítios regulatórios e orientar o desenho de fármacos que atuem não apenas bloqueando sítios ativos, mas reconfigurando as conversas internas que fazem as proteínas funcionarem.
Citação: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Palavras-chave: protease principal do SARS-CoV-2, alosteria, dinâmica de proteínas, cristalografia, descoberta de fármacos