Clear Sky Science · de
Statistische Kristallographie deckt ein allosterisches Netzwerk in SARS-CoV-2 Mpro auf
Wie Formwandel von Proteinen einem Coronavirus-Enzym hilft
Das Virus, das COVID-19 verursacht, ist auf eine molekulare Maschine angewiesen, die sogenannte Hauptprotease, um lange virale Ketten in funktionsfähige Teile zu zerschneiden. Medikamente richten sich bereits gegen dieses Enzym, doch es ist weiterhin schwierig nachzuvollziehen, wie seine feinen internen Bewegungen steuern, wann es ein- oder ausgeschaltet ist. In dieser Studie wurden mehr als tausend Momentaufnahmen der in Kristallen fixierten Protease verwendet, um zu zeigen, wie entfernte Bereiche des Moleküls mit dem Geschäftsbereich, in dem die Chemie stattfindet, kommunizieren — und wie diese verborgene Kommunikation gestört werden kann.
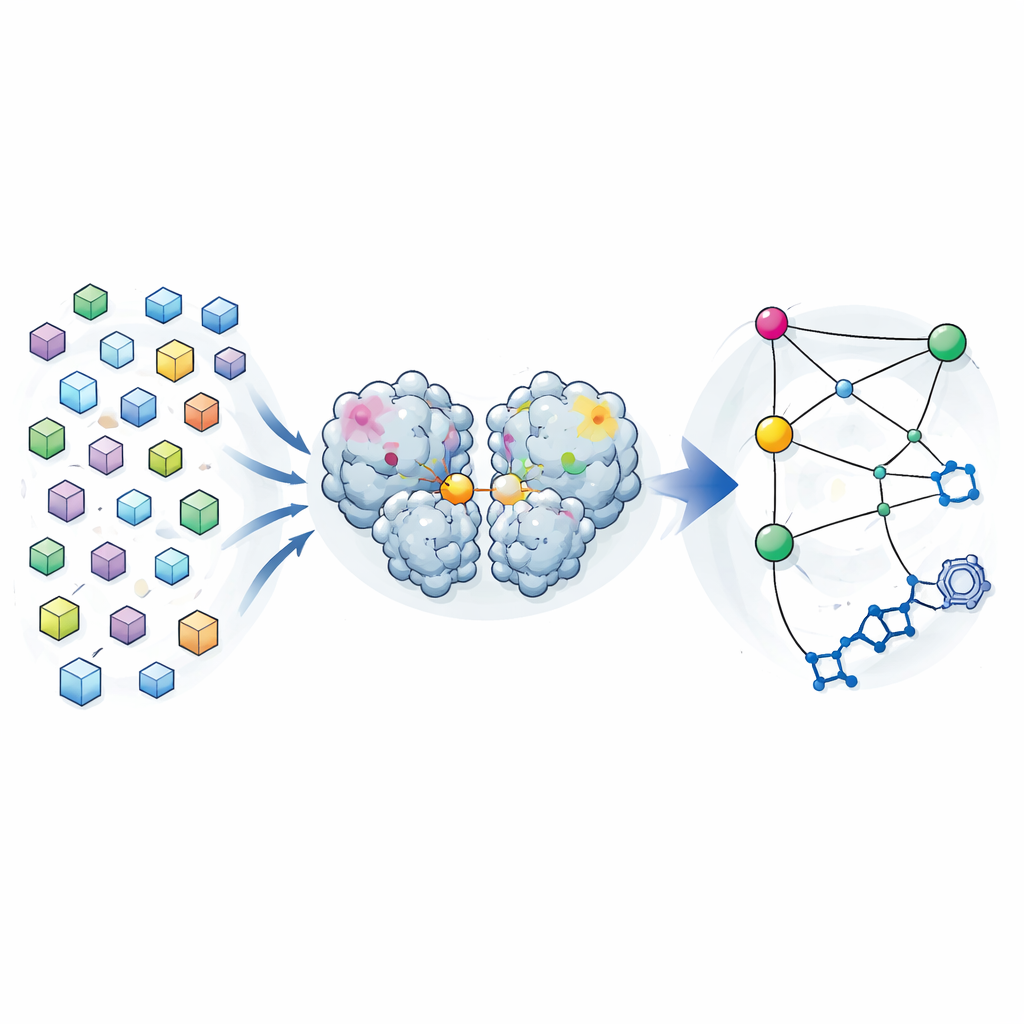
Viele Momentaufnahmen statt nur einer
Die traditionelle Kristallographie zielt darauf ab, eine einzelne „beste“ Struktur eines Proteins zu liefern, gemittelt über viele Moleküle im Kristall. Die Autoren kehrten diese Denkweise um. Während einer früheren Wirkstoff-Suchkampagne hatten sie Beugungsdaten von fast 7000 Kristallen der SARS-CoV-2-Hauptprotease gesammelt. Nach sorgfältigem Herausfiltern aller Kristalle mit gebundenen Verbindungen und unter strikter Qualitätskontrolle blieben 1146 hochaufgelöste Strukturen des Enzyms allein übrig. Statt kleine Unterschiede zwischen diesen Strukturen als bloßes Rauschen abzutun, analysierten sie diese als Verteilung von Formen, die die Protease einnehmen kann, während sie nahe an ihrer arbeitenden Konformation bleibt.
Langreichweitige Verbindungen innerhalb des Enzyms finden
Indem sie verglichen, wie sich die Position jedes Atoms von Struktur zu Struktur variierte, erstellte das Team eine Kovarianzkarte — eine Art statistischer Fingerabdruck, der zeigt, welche Regionen dazu neigen, sich gemeinsam zu bewegen. Sie fanden, dass diese auf Kristallen basierenden Korrelationen denjenigen ähnelten, die von langen Molekulardynamiksimulationen der Protease in Lösung vorhergesagt wurden. Das deutet darauf hin, dass die in den Kristallen eingefrorene Variabilität reale Schwingbewegungen widerspiegelt, die das Enzym in Wasser erkundet. Als sie untersuchten, wie verschiedene Regionen mit dem aktiven Zentrum — der Tasche, die das Schneiden übernimmt — korrelierten, entdeckten sie drei „Hotspots“ in der Dimerisierungsdomäne der Protease, dem Abschnitt, der zwei Exemplare des Enzyms beim Paaren unterstützt. Veränderungen an diesen Hotspots, weit entfernt vom aktiven Zentrum, schienen eng mit Veränderungen in der katalytischen Tasche verknüpft zu sein.
Die Hotspots durch gezielte Veränderungen testen
Um zu prüfen, ob diese statistisch identifizierten Stellen tatsächlich die Funktion kontrollieren, ersetzten die Forscher an jedem Hotspot eine einzelne Aminosäure durch Alanin, eine gebräuchliche Methode, um die lokale Struktur leicht zu verändern. Sie maßen dann, wie gut jedes Mutantenenzym Dimere bildete, ein substratähnliches Fragment band und ein fluoreszierendes Testpeptid schnitt. Eine Position, N214, verhielt sich als bestätigter „allosterischer Hotspot“: Die Mutation schwächte die Dimerbildung um mehr als den Faktor zehn, und selbst wenn genügend Dimere vorhanden waren, sank die katalytische Rate um etwa eine Größenordnung. Hochauflösende Strukturen des Mutanten zeigten den Grund: Das lokale Wasserstoffbrücknetzwerk, das normalerweise eine entscheidende Schleife stabilisiert, die das „Oxyanionloch“ im aktiven Zentrum bildet, wurde zerstört, und diese Schleife wurde teilweise ungeordnet und für die Chemie fehl ausgerichtet.
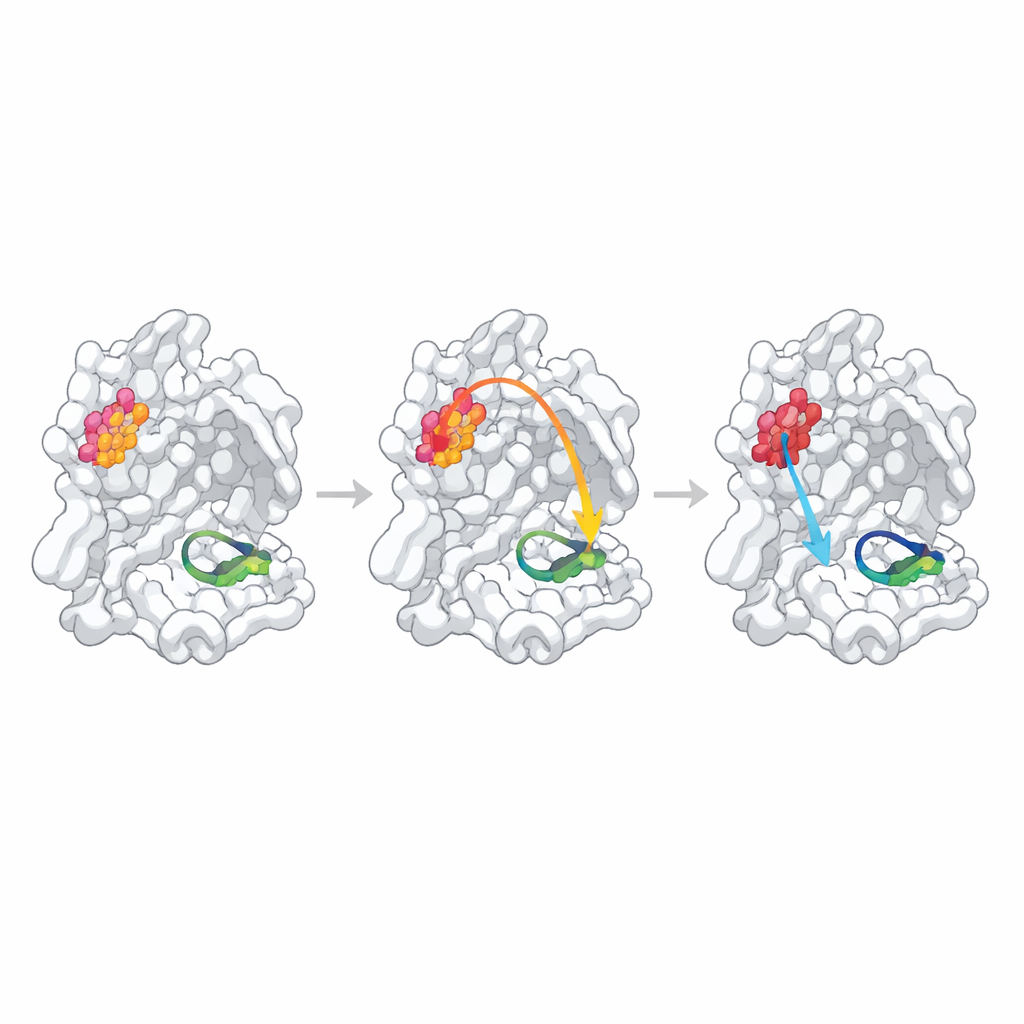
Wenn vorhergesagte Verbindungen schwach oder verborgen sind
Die beiden anderen vorhergesagten Hotspots verhalten sich anders. Die Veränderung von Q256, einer gegenüber Lösungsmittel exponierten und nicht offensichtlich an Dimerkontakten beteiligten Aminosäure, führte nur zu einem moderaten Aktivitätsabfall, der größtenteils durch etwas schwächere Dimerbildung und etwas schlechtere Bindung des Testinhibitors erklärt werden kann. Seine Kristallstruktur zeigte eine völlig neue Packungsanordnung, in der benachbarte Moleküle die durch die Mutation entstandene Höhlung füllten, wodurch es schwer wurde, echte interne Kommunikation von kristallspezifischen Effekten zu unterscheiden. Die dritte Stelle, S284, liegt in einem kleinen „Reißverschluss“, der die beiden Enzymkopien verbindet. Wie aus Sequenzvergleichen mit verwandten Coronaviren zu erwarten war, hatte der Austausch dieser Aminosäure nahezu keinen Effekt auf Struktur oder Funktion, was darauf hinweist, dass diese spezielle Alanin-Substitution eine zu milde Störung war, um den vorhergesagten Pfad zu testen.
Kristalle als winzige Kraftexperimente
Ein interessantes Nebenergebnis der Arbeit ist, dass viele der strukturellen Unterschiede zwischen Kristallen auf winzige Veränderungen im Kristallgitter — der sich wiederholenden Anordnung von Proteinen und Lösungsmittel — zurückgeführt werden konnten. Während Kristalle beim Umgang leicht unterschiedlich austrockneten, schrumpften oder dehnten sich ihre Einheitszellen, und diese Verschiebungen zogen an Kontaktpunkten zwischen benachbarten Proteasemolekülen. Diese Kräfte pflanzten sich durch das Protein fort und erzeugten die strukturelle Vielfalt, die der Kovarianzanalyse zugrunde lag. Mit anderen Worten: Jeder Kristall fungierte als kleines mechanisches Experiment, das die Protease sanft in verschiedene Richtungen spannte und zeigte, wie sie sich biegt, ohne auseinanderzufallen.
Warum das für Biologie und Wirkstoffdesign wichtig ist
Indem die Autoren Tausende von Kristallstrukturen als statistische Population statt als einzelne Antwort behandelten, konnten sie ein allosterisches Netzwerk ableiten, das eine Dimeroberflächen-Residue, N214, mit dem katalytischen Zentrum der SARS-CoV-2-Hauptprotease verbindet. Die Störung dieser Stelle reduziert nicht einfach die Anzahl gebildeter Dimere; sie verwirrt spezifisch die Geometrie der aktiven Schleife, die für effiziente Katalyse erforderlich ist. Dies bestätigt, dass die Aktivität des Enzyms durch fernwirkende Kommunikation über das ganze Protein gesteuert wird. Allgemeiner demonstriert die Studie einen Ansatz der „statistischen Kristallographie“, bei dem unkontrollierte, alltägliche Störungen von Kristallen — etwa geringe Unterschiede im Austrocknen — zu einer Ressource werden, um funktionelle Bewegungen zu kartieren. Wenn ähnliche Datensätze für andere Proteine gesammelt werden, könnte diese Strategie helfen, neue Regulationsstellen aufzudecken und das Design von Wirkstoffen zu leiten, die nicht nur aktive Zentren blockieren, sondern die internen Kommunikationswege umgestalten, die Proteine antreiben.
Zitation: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Schlüsselwörter: SARS-CoV-2 Hauptprotease, Allosterie, Proteindynamik, Kristallographie, Arzneimittelentdeckung