Clear Sky Science · it
La cristallografia statistica rivela una rete allosterica nella Mpro di SARS-CoV-2
Come il cambiamento di forma delle proteine aiuta a funzionare un enzima del coronavirus
Il virus che causa il COVID-19 dipende da una macchina molecolare chiamata proteasi principale per tagliare lunghe catene virali in parti funzionanti. I farmaci mirano già a quest’enzima, ma gli scienziati faticano ancora a capire come i suoi sottili movimenti interni controllino l’accensione o lo spegnimento dell’attività. Questo studio utilizza più di mille istantanee della proteasi bloccata in cristalli per rivelare come regioni distanti della molecola comunicano con il centro in cui avviene la chimica — e mostra come questa comunicazione nascosta possa essere interrotta.
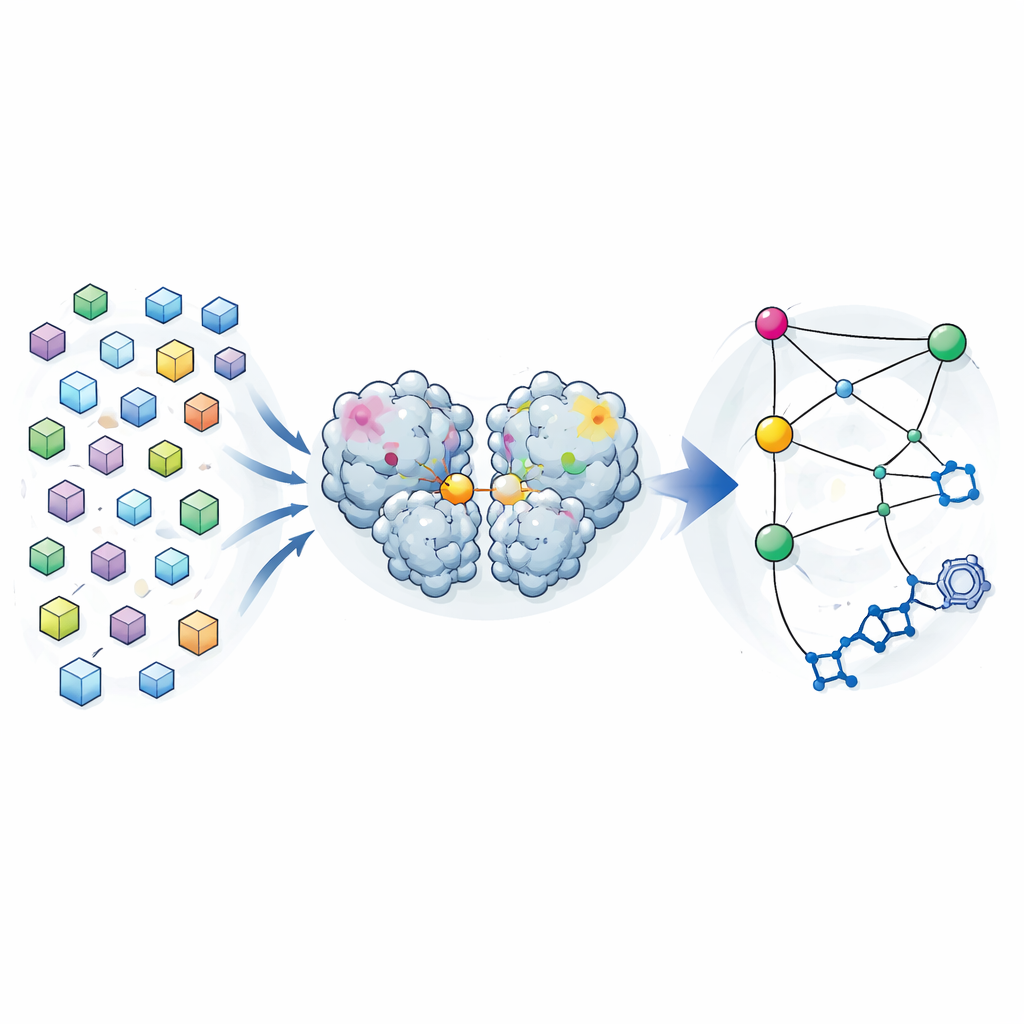
Molte istantanee invece di una sola
La cristallografia tradizionale mira a fornire una singola “migliore” struttura di una proteina, mediata su molte molecole in un cristallo. Gli autori hanno rovesciato questa logica. Durante una precedente campagna di ricerca di farmaci, avevano raccolto dati di diffrazione da quasi 7000 cristalli della proteasi principale di SARS-CoV-2. Dopo aver attentamente filtrato i cristalli che contenevano composti legati e aver applicato limiti qualitativi rigorosi, hanno ottenuto 1146 strutture ad alta risoluzione dell’enzima isolato. Piuttosto che trattare le piccole differenze tra queste strutture come semplice rumore, le hanno analizzate come una distribuzione di forme che la proteasi può adottare rimanendo vicina alla sua forma funzionante.
Trovare connessioni a lunga distanza all’interno dell’enzima
Confrontando come la posizione di ogni atomo variava da struttura a struttura, il team ha costruito una mappa di covarianza — una sorta di impronta statistica che mostra quali regioni tendono a muoversi insieme. Hanno scoperto che queste correlazioni basate sui cristalli somigliavano molto a quelle previste da lunghe simulazioni di dinamica molecolare della proteasi in soluzione. Ciò suggerisce che la variabilità congelata nei cristalli riflette reali movimenti di flessione che l’enzima esplora in acqua. Quando si sono concentrati su come diverse regioni si correllavano con il sito attivo — la tasca che effettua il taglio — hanno identificato tre “hotspot” nel dominio di dimerizzazione della proteasi, la porzione che aiuta due copie dell’enzima ad accoppiarsi. Le variazioni in questi hotspot, lontani dal sito attivo, sembravano strettamente collegate a cambiamenti nella tasca catalitica.
Testare gli hotspot con modifiche mirate
Per sondare se questi siti identificati statisticamente controllassero davvero la funzione, i ricercatori hanno alterato ciascun hotspot sostituendo un singolo amminoacido con alanina, un modo standard per perturbare la struttura locale. Hanno quindi misurato quanto bene ogni proteasi mutante formava dimeri, legava un frammento simile al substrato e tagliava un peptide test fluorescente. Una posizione, N214, si è comportata come un “hotspot allosterico” confermato: la mutazione ha indebolito la formazione dei dimeri di oltre dieci volte e, anche quando erano presenti dimeri in numero sufficiente, la velocità catalitica è diminuita di circa un ordine di grandezza. Le strutture ad alta risoluzione del mutante hanno spiegato il perché: la rete locale di legami a idrogeno che normalmente stabilizza un’ansa cruciale che forma il “foro ossianionico” nel sito attivo è stata interrotta, e quest’ansa è diventata parzialmente disordinata e mal allineata per la chimica.
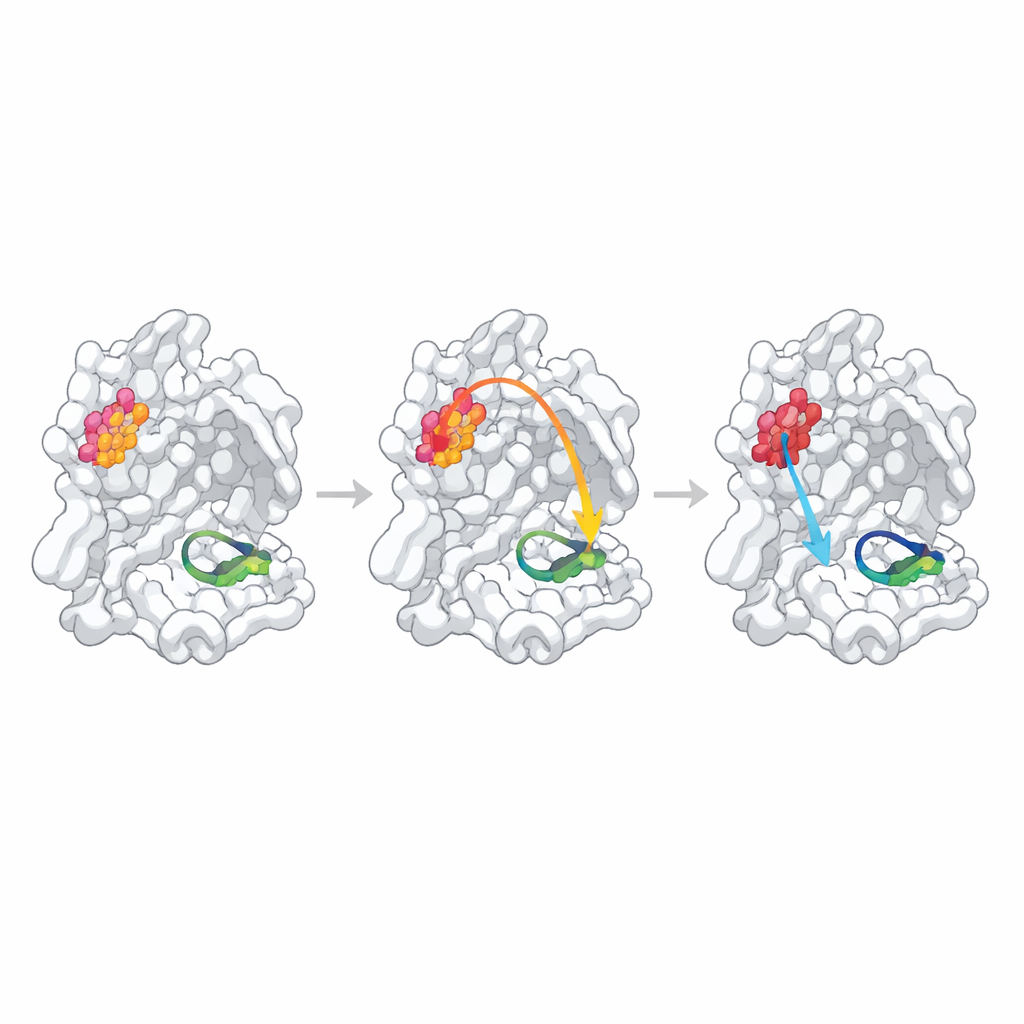
Quando i legami previsti sono deboli o nascosti
Gli altri due hotspot previsti si sono comportati in modo diverso. La sostituzione di Q256, un residuo esposto al solvente e non evidentemente coinvolto nei contatti di dimerizzazione, ha prodotto solo una modesta diminuzione dell’attività spiegabile in gran parte con una formazione di dimeri leggermente più debole e un legame un po’ peggiore dell’inibitore di prova. La sua struttura cristallina ha rivelato un nuovo assetto di impaccamento in cui molecole vicine hanno riempito la cavità lasciata dalla mutazione, rendendo difficile distinguere la vera comunicazione interna da effetti specifici del cristallo. Il terzo sito, S284, si trova in una piccola “cerniera” che collega le due copie dell’enzima. Come previsto dal confronto delle sequenze con coronavirus correlati, la sostituzione di questo residuo ha avuto quasi nessun effetto su struttura o funzione, indicando che questa particolare sostituzione con alanina è stata una perturbazione troppo lieve per testare il percorso previsto.
I cristalli come piccoli esperimenti di forza
Un risultato laterale interessante del lavoro è che molte delle differenze strutturali tra i cristalli potevano essere ricondotte a piccole variazioni nel reticolo cristallino — l’ordine ripetuto di proteine e solvente. Man mano che i cristalli si asciugavano in misura leggermente diversa durante la manipolazione, le loro celle unitarie si restringevano o si allungavano, e questi spostamenti tiravano i punti di contatto tra molecole di proteasi adiacenti. Quelle forze si propagavano attraverso la proteina, generando la diversità strutturale che ha sostenuto l’analisi di covarianza. In altre parole, ogni cristallo ha agito come un piccolo esperimento meccanico, sollecitando delicatamente la proteasi in direzioni diverse e rivelando come si flette senza disgregarsi.
Perché questo conta per la biologia e la progettazione di farmaci
Trattando migliaia di strutture cristalline come una popolazione statistica invece che come una singola risposta, gli autori hanno potuto inferire una rete allosterica che collega un residuo dell’interfaccia di dimerizzazione, N214, al centro catalitico della proteasi principale di SARS-CoV-2. Interrompere questo sito non si limita a ridurre il numero di dimeri formati; sconvolge specificamente la geometria dell’ansa attiva necessaria per una catalisi efficiente. Questo conferma che l’attività dell’enzima è governata da comunicazioni a lunga distanza attraverso la proteina. Più in generale, lo studio dimostra un approccio di “cristallografia statistica” in cui perturbazioni incontrollate e quotidiane dei cristalli — come lievi differenze nell’essiccazione — diventano una risorsa per mappare i moti funzionali. Man mano che dataset simili verranno raccolti per altre proteine, questa strategia potrebbe aiutare a scoprire nuovi siti regolatori e guidare la progettazione di farmaci che agiscono non solo bloccando i siti attivi ma rimodulando le conversazioni interne che fanno funzionare le proteine.
Citazione: Creon, A., Scheer, T.E.S., Reinke, P. et al. Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro. Commun Biol 9, 602 (2026). https://doi.org/10.1038/s42003-026-10127-w
Parole chiave: proteasi principale di SARS-CoV-2, allosteria, dinamica delle proteine, cristallografia, scoperta di farmaci