Clear Sky Science · sv
En hotspot-fosforyleringsplats på SHP2 driver onkoproteinaktivering och läkemedelsresistens
Varför detta spelar roll för framtida cancerbehandlingar
Många moderna cancerläkemedel syftar till att stänga av överaktiva tillväxtsignaler inne i tumörceller. Ett lovande mål har varit proteinet SHP2, en viktig brytare som kopplar signaler vid cellens yta till cellens inre tillväxtmotor. Ändå har läkemedel utformade för att stänga ner SHP2 visat nästan ingen nytta i tidiga kliniska prövningar. Denna artikel avslöjar en dold "på-knapp" på SHP2 som hjälper till att förklara varför dessa läkemedel ofta misslyckas och pekar på nya sätt att överlista resistenta tumörer.
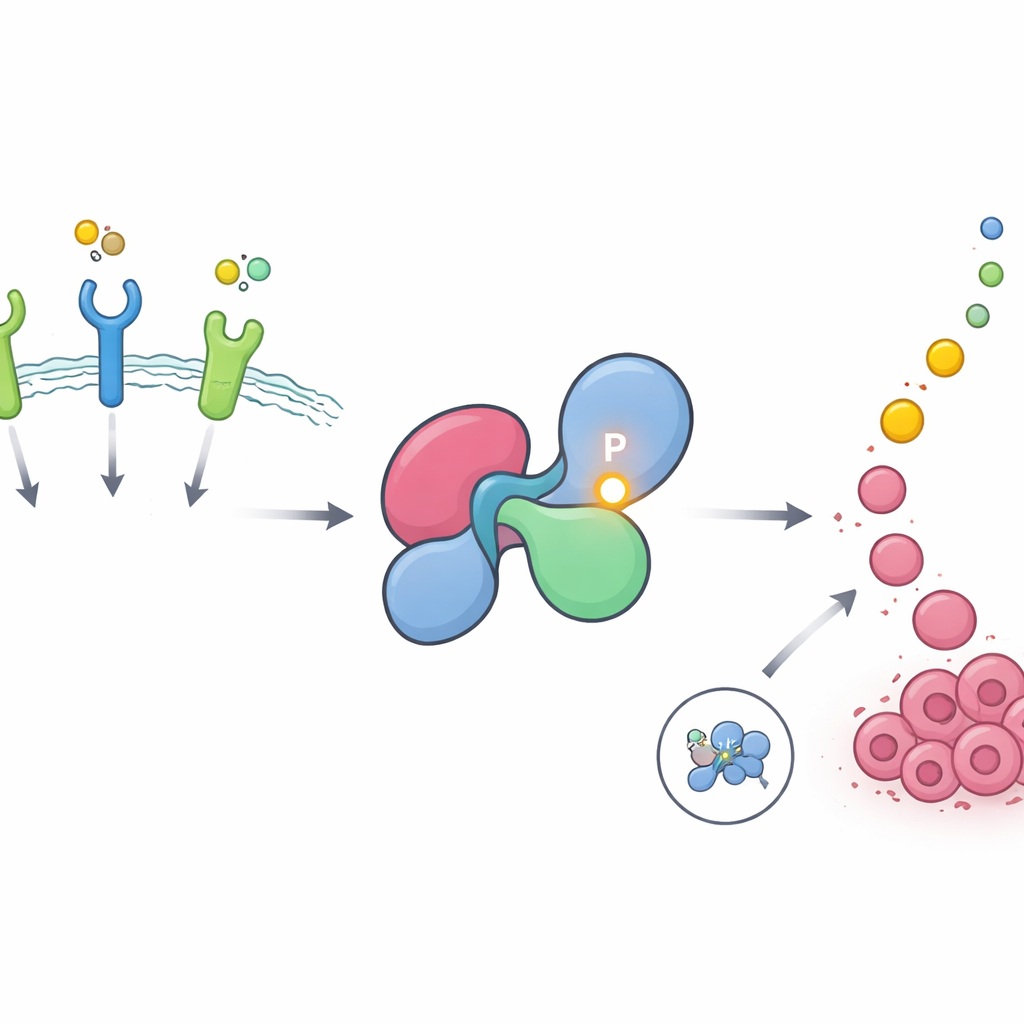
En dold hotspot på en avgörande tillväxtbrytare
SHP2 hjälper till att förmedla meddelanden från receptorer på cellens yta, så kallade receptor-tyrosinkinaser, till RAS–MAPK‑vägen, en huvuddrivkraft för celltillväxt och delning. I friska celler är SHP2 mestadels inaktivt, vikt i en sluten form som blockerar det egna aktiva centret. Det öppnar sig kort när receptorer stimuleras, för att sedan stängas igen. Författarna använde stora fosfoproteomiska databaser för att söka efter proteinplatser som ofta modifieras genom tillsats av fosfatgrupper, ett vanligt sätt för celler att slå signaler på och av. De fann att en särskild position på SHP2, tyrosin 62, är bland de mest intensivt och återkommande fosforylerade platserna i hela det mänskliga proteomet och är särskilt rikligt förekommande i tumörer drivna av tillväxtfaktorreceptorer som EGFR och FGFR.
Olika cancerformer, olika beroende av samma brytare
Genom att gräva i patienttumördatasets visade forskarna att fosforylering vid SHP2:s Y62-plats är starkt ökande i flera receptor-drivna cancerformer, inklusive vissa lungcancer, gallgångscancer, huvud- och halsstumörer, samt hjärntumörer med EGFR‑förändringar. Slående var att i vissa lungcancerformer var SHP2 Y62-fosforylering ännu högre än fosforylering på receptorn själv. I kontrast visade tumörer drivna av muterat KRAS, som aktiverar samma tillväxtväg nedströms, minskade nivåer av SHP2 Y62-fosforylering. Detta mönster tyder på att cancerformer som förlitar sig på receptorsignaler ofta förstärker SHP2 Y62 som en del av sin koppling, medan de med mutationer längre ner i vägen inte behöver just denna brytare.
Hur en annan kinasfamilj kontrollerar denna hotspot
I ett första skede var det naturligt att misstänka att ytreceptorerna själva direkt modifierar SHP2 vid Y62. När teamet blockerade olika receptorer med riktade läkemedel förändrades dock Y62-fosforyleringen knappt. Istället följde de modifieringen till en annan grupp enzymer kända som SRC-familjens kinaser. Med en kombination av breda och selektiva SRC‑familjsinhibitorer, genetiska knockoutceller utan SRC, YES1 och FYN, samt renade proteiner i provrörsreaktioner, visade de att dessa kinaser direkt tillsätter fosfatgrupper på SHP2 vid Y62, och också på två bättre kända platser på SHP2:s svans. Detta placerar SRC‑familjens kinaser som kritiska mellanled som sitter mellan aktiverade receptorer och SHP2 och skapar en RTK–SRC–SHP2‑signalaxel.
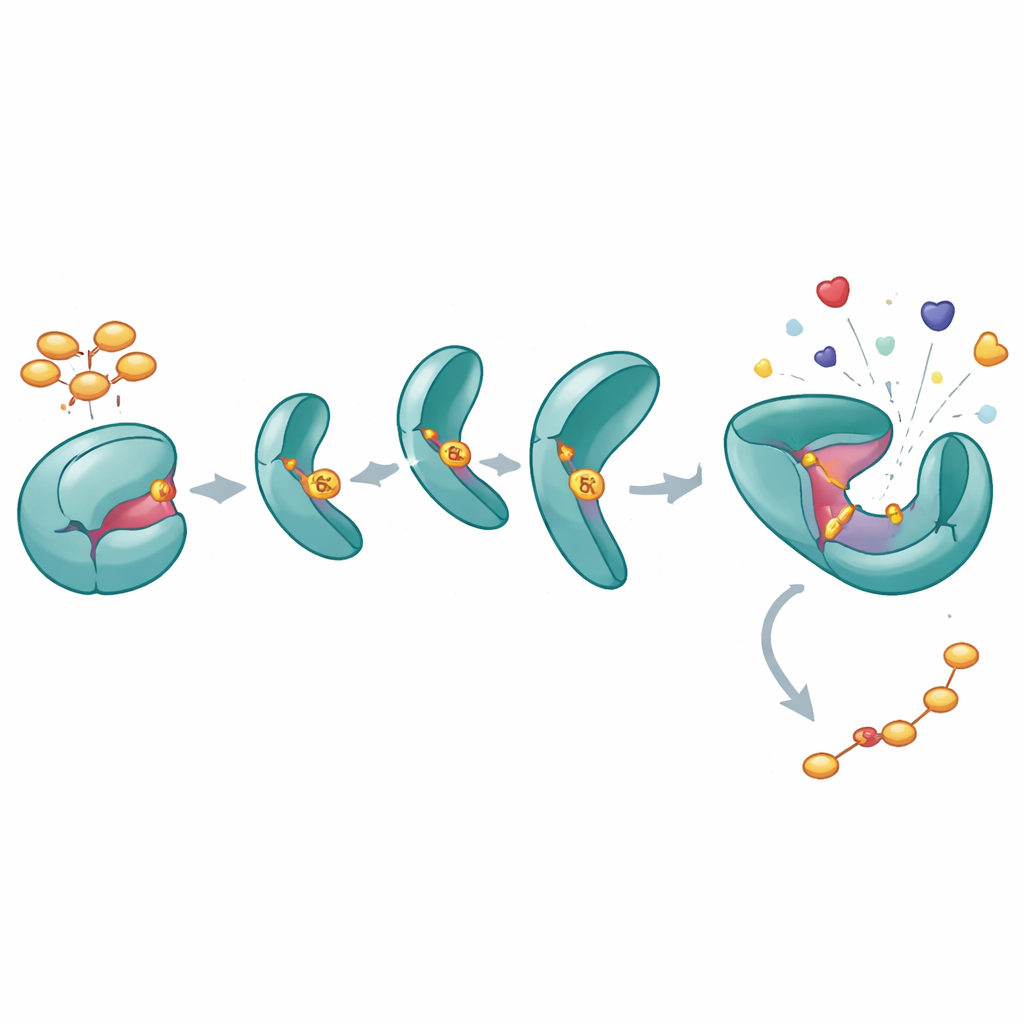
Hur en fosfatgrupp håller SHP2 vidöppet
För att förstå vad Y62‑fosforylering gör med SHP2 konstruerade författarna en "fosfomimetisk" variant (Y62D) som beter sig som om Y62 var permanent fosforylerad. Biokemiska tester visade att denna variant har högre katalytisk aktivitet och svarar inte normalt på aktiverande peptider som vanligtvis skulle "låsa upp" SHP2 från dess slutna tillstånd. Mätningar av termisk stabilitet och väte–deuterium‑utbytesexperiment visade att Y62D‑proteinet provar mer öppna, flexibla konformationer, särskilt vid gränssnittet mellan dess regulatoriska och katalytiska domäner. Medan en kristallstruktur av Y62D fångade den i en sluten form, indikerade de dynamiska mätningarna att denna mutant i lösning ofta flippas till ett öppet, aktiv-liknande tillstånd och därigenom kringgår normal autoinhibering.
Varför nuvarande SHP2‑läkemedel har svårt i kliniken
Nuvarande allosteriska SHP2‑hämmare fungerar genom att stabilisera den slutna, inaktiva formen av SHP2, effektivt genom att "limma" dess domäner tillsammans. Författarna visade att Y62D‑formen av SHP2 är mindre känslig för dessa läkemedel i renade proteinassay. I cancercellinjer där det normala SHP2‑genen ersatts med Y62D förblev tillväxtsignaleringen via MAPK‑vägen hög även när cellerna behandlades med tre olika SHP2‑hämmare i klinisk fas. Dessa celler bibehöll också fosforylering av GAB1, en viktig signaladapter som är beroende av SHP2, vilket lyfter fram att SHP2 förblev funktionellt aktivt trots läkemedelsbehandling. Tillsammans stöder data en modell där SRC‑familjens kinaser fosforylerar SHP2 vid Y62 och tvingar det till en öppen, aktiv konformation som befintliga läkemedel inte effektivt kan låsa ner.
Vad detta betyder för morgondagens cancerterapier
Studien identifierar SHP2 Y62‑fosforylering som en vanlig, läkemedelsrelevant brytare som imiterar effekten av permanenta aktiverande mutationer, men utan förändringar i DNA:t. Detta hjälper till att förklara varför tumörer drivna av tillväxtfaktorreceptorer kan vara intrinsikt resistenta mot nuvarande SHP2‑hämmare, även om deras SHP2‑gen ser normal ut i sekvenseringstester. Att mäta Y62‑fosforylering kan fungera som en biomarkör för att förutsäga vilka tumörer som troligen inte svarar på dessa läkemedel. Viktigare är att arbetet framhäver Y62‑fosforylerat SHP2 som ett särskilt strukturellt mål och antyder att effektiva strategier kan kräva antingen att blockera SRC‑familjens kinaser uppströms, eller att utforma nya SHP2‑hämmare som direkt känner igen och stänger ner dess öppna, fosforylerade form.
Citering: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Nyckelord: SHP2, fosforylering, SRC-kinaser, MAPK-signalering, läkemedelsresistens