Clear Sky Science · de
Eine Hotspot-Phosphorylierungsstelle auf SHP2 treibt Onkoprotein‑Aktivierung und Arzneimittelresistenz
Warum das für künftige Krebstherapien wichtig ist
Viele moderne Krebstherapeutika zielen darauf ab, überaktive Wachstumssignale innerhalb von Tumorzellen auszuschalten. Ein vielversprechendes Ziel ist das Protein SHP2, ein zentraler Schalter, der Signale von der Zelloberfläche mit dem inneren Wachstums‑Apparat der Zelle verbindet. Dennoch zeigten Wirkstoffe, die SHP2 abschalten sollen, in frühen klinischen Studien kaum Nutzen. Diese Arbeit deckt einen verborgenen „Ein‑Schalter“ auf SHP2 auf, der erklärt, warum diese Medikamente oft versagen, und weist auf neue Wege hin, resistente Tumoren zu umgehen.
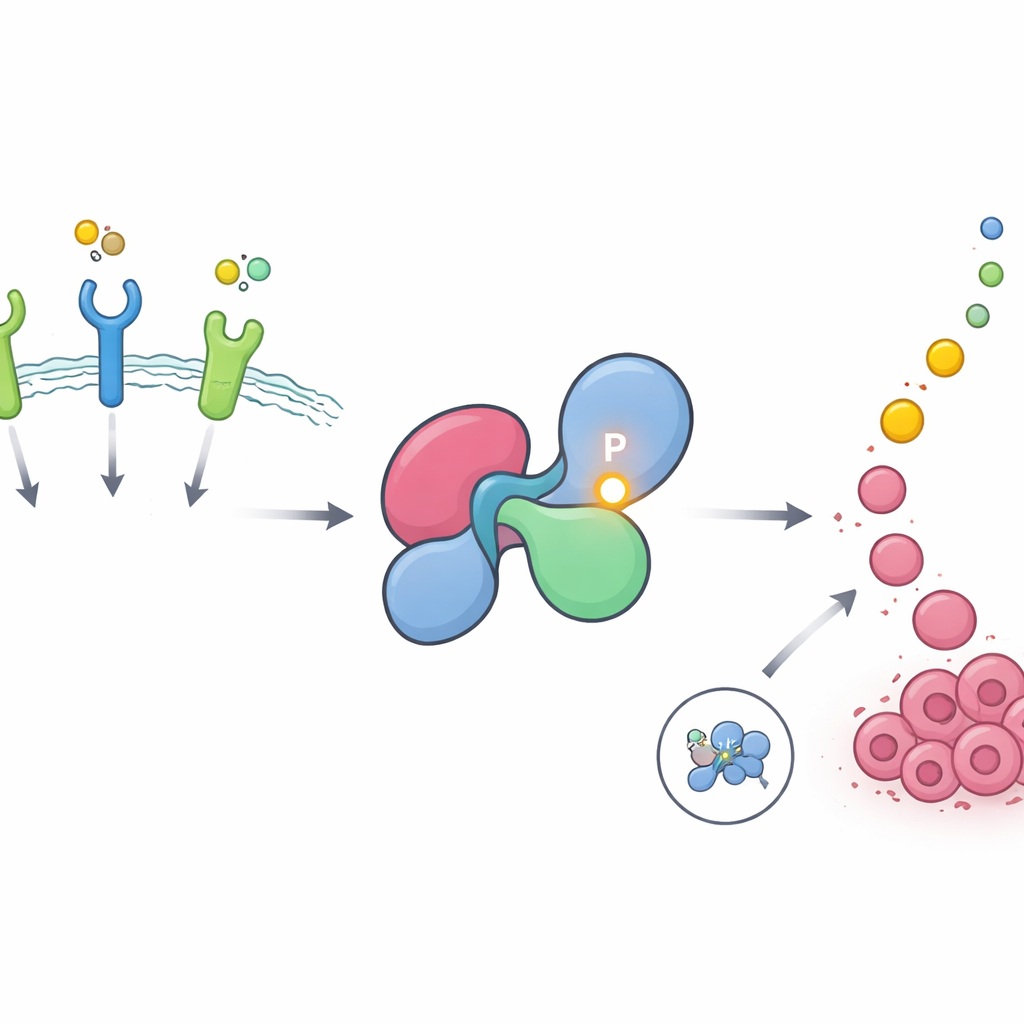
Ein verborgener Hotspot an einem zentralen Wachstumsschalter
SHP2 vermittelt Nachrichten von Rezeptoren an der Zelloberfläche, sogenannten Rezeptor‑Tyrosinkinasen, an den RAS–MAPK‑Signalweg, einen Haupttreiber von Zellwachstum und Zellteilung. In gesunden Zellen ist SHP2 überwiegend inaktiv und zu einer geschlossenen Form gefaltet, die die eigene aktive Stelle blockiert. Es öffnet sich kurz, wenn Rezeptoren stimuliert werden, und schließt sich dann wieder. Die Autorinnen und Autoren nutzten umfangreiche Phosphoproteom‑Datenbanken, um Proteinstellen zu finden, die häufig durch Zugabe von Phosphatgruppen verändert werden, einen üblichen Mechanismus zur Ein‑ und Ausschaltung zellulärer Signale. Sie fanden, dass eine bestimmte Position auf SHP2, Tyrosin 62 genannt, zu den am stärksten und wiederkehrend phosphorylierten Stellen im gesamten menschlichen Proteom gehört und insbesondere in Tumoren angereichert ist, die durch Wachstumsfaktorrezeptoren wie EGFR und FGFR angetrieben werden.
Verschiedene Krebsarten, unterschiedliche Abhängigkeit vom gleichen Schalter
Durch die Auswertung von Patiententumor‑Datensätzen zeigten die Forschenden, dass die Phosphorylierung an der Y62‑Stelle von SHP2 in mehreren rezeptorgetriebenen Krebserkrankungen deutlich erhöht ist, darunter bestimmte Lungenkrebse, Gallengangskrebs, Kopf‑ und Hals‑Tumoren sowie Gehirntumoren mit EGFR‑Veränderungen. Bemerkenswerterweise war in einigen Lungenkrebstypen die SHP2‑Y62‑Phosphorylierung sogar stärker erhöht als die Phosphorylierung des Rezeptors selbst. Im Gegensatz dazu zeigten Tumoren, die von mutiertem KRAS angetrieben werden – das denselben Wachstumsweg weiter stromab aktiviert –, verringerte SHP2‑Y62‑Phosphorylierungswerte. Dieses Muster legt nahe, dass Krebsarten, die auf Rezeptorsignale angewiesen sind, häufig SHP2‑Y62 als Teil ihrer Signalverkabelung hochregulieren, während solche mit weiter unten wirkenden Mutationen diesen speziellen Schalter nicht benötigen.
Wie eine andere Kinase‑Familie diesen Hotspot kontrolliert
Auf den ersten Blick lag es nahe zu vermuten, dass die Oberflächenrezeptoren selbst SHP2 an Y62 direkt modifizieren. Als das Team jedoch verschiedene Rezeptoren mit gezielten Wirkstoffen blockierte, veränderte sich die Y62‑Phosphorylierung kaum. Stattdessen führten ihre Spurensuchen zu einer anderen Gruppe von Enzymen, den SRC‑Familienkinasen. Durch eine Kombination aus breit und selektiv wirkenden SRC‑Familieninhibitoren, genetischen Knockout‑Zellen ohne SRC, YES1 und FYN sowie Reaktionen mit gereinigten Proteinen zeigten sie, dass diese Kinasen SHP2 direkt an Y62 phosphorylieren und zudem an zwei bekannteren Stellen am C‑Terminal von SHP2. Damit nehmen SRC‑Familienkinasen eine zentrale Vermittlerrolle zwischen aktivierten Rezeptoren und SHP2 ein und bilden eine RTK–SRC–SHP2‑Signalkaskade.
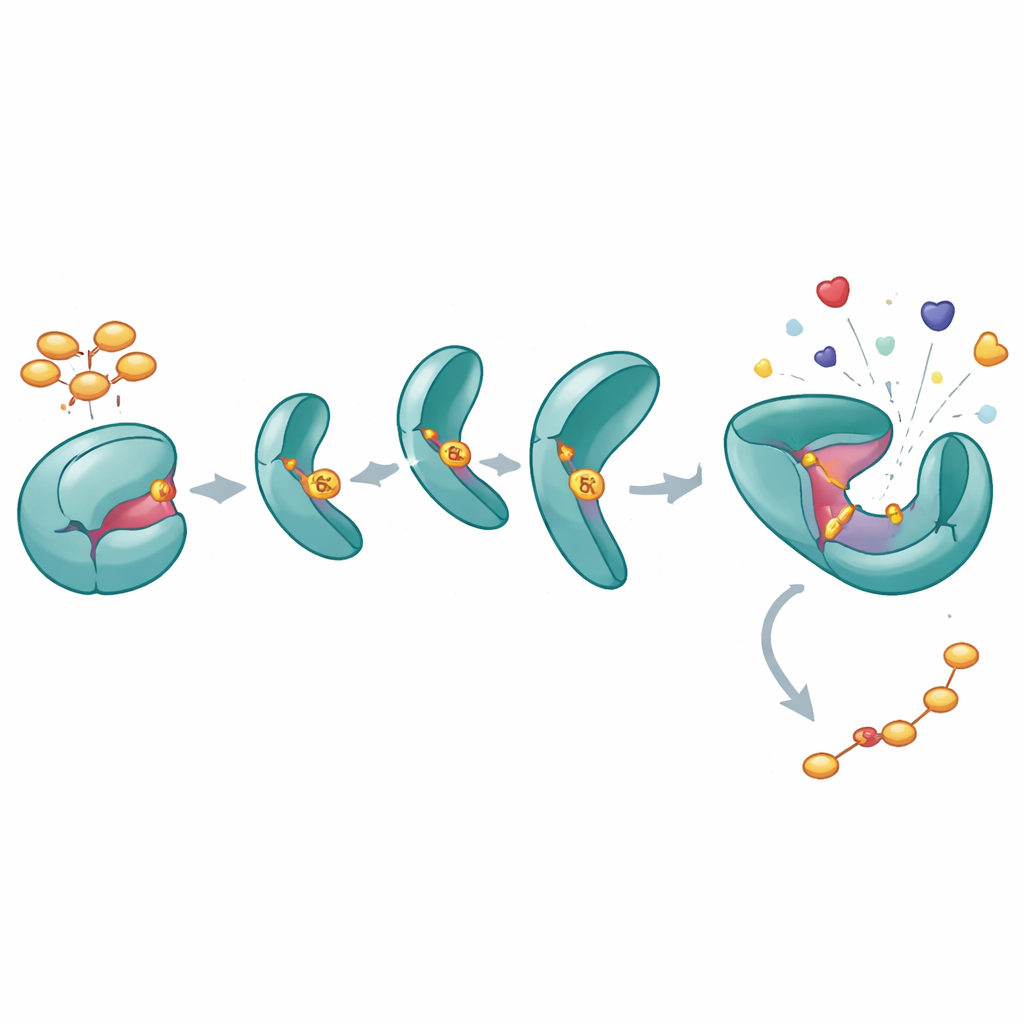
Wie eine einzige Phosphatgruppe SHP2 dauerhaft offenhält
Um zu verstehen, welche Auswirkungen die Y62‑Phosphorylierung auf SHP2 hat, konstruierten die Autorinnen und Autoren eine „phosphomimetische“ Variante (Y62D), die sich so verhält, als wäre Y62 dauerhaft phosphoryliert. Biochemische Assays zeigten, dass diese Variante eine höhere katalytische Aktivität besitzt und nicht normal auf aktivierende Peptide reagiert, die typischerweise SHP2 aus seinem geschlossenen Zustand „entsperren“. Messungen der thermischen Stabilität und Wasserstoff‑Deuterium‑Austausch‑Experimente offenbarten, dass das Y62D‑Protein häufiger offenere, flexiblere Konformationen einnimmt, insbesondere an der Schnittstelle zwischen seinen regulatorischen und katalytischen Domänen. Während eine Kristallstruktur von Y62D eine geschlossene Form einfing, deuteten die dynamischen Messungen darauf hin, dass dieses Mutant in Lösung häufig in einen offenen, aktivitätsähnlichen Zustand umschlägt und so die normale Auto‑Inhibition umgeht.
Warum aktuelle SHP2‑Medikamente klinisch schwer durchschlagen
Aktuelle allosterische SHP2‑Inhibitoren wirken, indem sie die geschlossene, inaktive Form von SHP2 stabilisieren und seine Domänen praktisch „verkleben“. Die Autorinnen und Autoren zeigten, dass die Y62D‑Form von SHP2 in gereinigten Proteinversuchen weniger empfindlich gegenüber diesen Wirkstoffen ist. In Krebszelllinien, in denen das normale SHP2‑Gen durch Y62D ersetzt wurde, blieb die Signalübertragung über den MAPK‑Weg hoch, selbst wenn die Zellen mit drei verschiedenen klinisch entwickelten SHP2‑Inhibitoren behandelt wurden. Diese Zellen behielten zudem die Phosphorylierung von GAB1 bei, einem wichtigen Signaladapter, der von SHP2 abhängt, was zeigt, dass SHP2 funktionell aktiv blieb, trotz Medikamentenbehandlung. Zusammengenommen stützen die Daten ein Modell, in dem SRC‑Familienkinasen SHP2 an Y62 phosphorylieren und es in eine offene, aktive Konformation zwingen, die von bestehenden Wirkstoffen nicht effizient fixiert werden kann.
Was das für die Therapien von morgen bedeutet
Die Studie identifiziert die SHP2‑Y62‑Phosphorylierung als einen häufigen, medikamentenrelevanten Schalter, der die Wirkung permanenter aktivierender Mutationen nachahmt, jedoch ohne Änderungen an der DNA. Das erklärt, warum durch Wachstumsfaktorrezeptoren getriebene Tumoren intrinsisch gegenwärtigen SHP2‑Inhibitoren widerstehen können, selbst wenn das SHP2‑Gen in Sequenztests normal erscheint. Die Messung der Y62‑Phosphorylierung könnte als Biomarker dienen, um vorherzusagen, welche Tumoren wahrscheinlich nicht auf diese Wirkstoffe ansprechen. Noch wichtiger macht die Arbeit SHP2 mit phosphoryliertem Y62 zu einem eigenständigen strukturellen Ziel und legt nahe, dass wirksame Strategien entweder die Blockade von SRC‑Familienkinasen stromaufwärts erfordern oder neue SHP2‑Inhibitoren, die die offene, phosphorylierte Form gezielt erkennen und abschalten, entwickelt werden müssen.
Zitation: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Schlüsselwörter: SHP2, Phosphorylierung, SRC‑Kinasen, MAPK‑Signalübertragung, Arzneimittelresistenz