Clear Sky Science · en
A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance
Why this matters for future cancer treatments
Many modern cancer drugs aim to turn off overactive growth signals inside tumor cells. One promising target has been a protein called SHP2, a key switch that connects signals at the cell surface to the cell’s internal growth engine. Yet drugs designed to shut down SHP2 have shown almost no benefit in early clinical trials. This paper uncovers a hidden “on switch” on SHP2 that helps explain why these drugs often fail and points to new ways to outsmart resistant tumors.
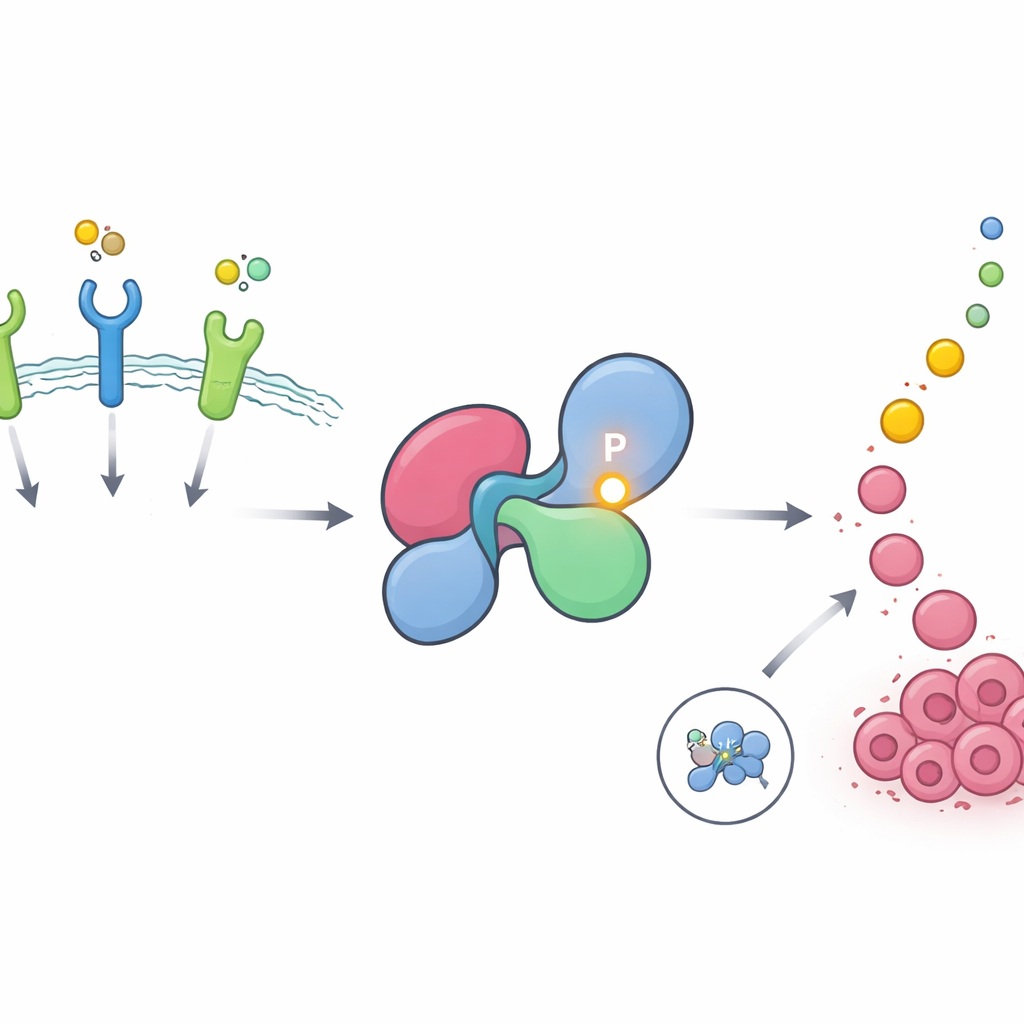
A hidden hot spot on a crucial growth switch
SHP2 helps relay messages from receptors on the cell surface, called receptor tyrosine kinases, to the RAS–MAPK pathway, a major driver of cell growth and division. In healthy cells, SHP2 stays mostly off, folded into a closed shape that blocks its own active site. It briefly opens when receptors are stimulated, then shuts again. The authors used large phosphoproteomic databases to search for protein sites that are frequently modified by the addition of phosphate groups, a common way cells turn signals on and off. They found that one particular position on SHP2, called tyrosine 62, is among the most heavily and recurrently phosphorylated sites in the entire human proteome and is especially enriched in tumors driven by growth factor receptors such as EGFR and FGFR.
Different cancers, different reliance on the same switch
By mining patient tumor datasets, the researchers showed that phosphorylation at SHP2’s Y62 site is strongly increased in several receptor-driven cancers, including certain lung cancers, bile duct cancers, head and neck tumors, and brain tumors with EGFR alterations. Strikingly, in some lung cancers, SHP2 Y62 phosphorylation was even more elevated than phosphorylation on the receptor itself. In contrast, tumors driven by mutant KRAS, which activates the same growth pathway downstream, showed reduced levels of SHP2 Y62 phosphorylation. This pattern suggests that cancers relying on receptor signals often boost SHP2 Y62 as part of their wiring, while those with mutations further down the pathway do not need this particular switch.
How another kinase family controls this hot spot
At first glance, it was natural to suspect that the surface receptors themselves directly modify SHP2 at Y62. However, when the team blocked various receptors with targeted drugs, Y62 phosphorylation barely budged. Instead, they traced the modification to a different group of enzymes known as SRC family kinases. Using a combination of broad and selective SRC-family inhibitors, genetic knockout cells lacking SRC, YES1, and FYN, and purified proteins in test-tube reactions, they showed that these kinases directly add phosphate groups to SHP2 at Y62, and also at two better-known sites on SHP2’s tail. This places SRC-family kinases as critical middlemen that sit between activated receptors and SHP2, creating an RTK–SRC–SHP2 signaling axis.
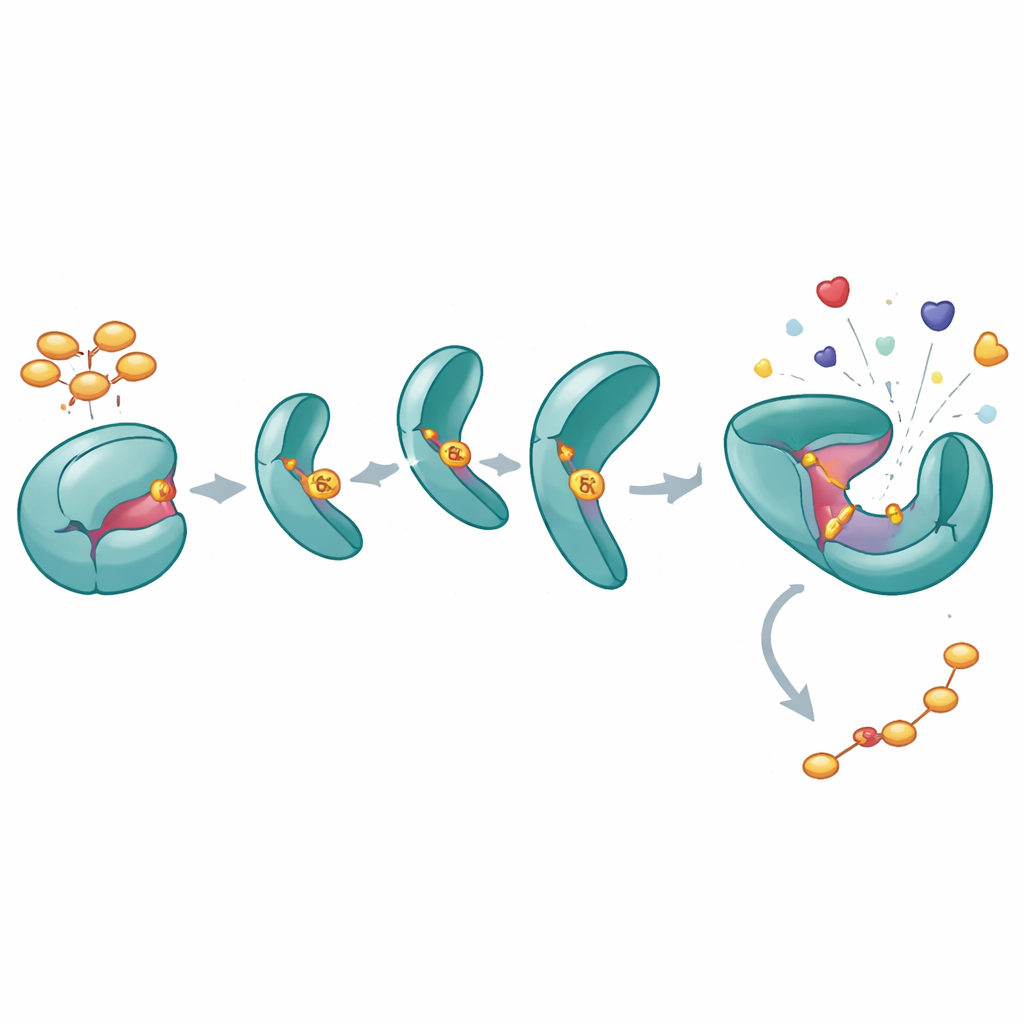
How one phosphate holds SHP2 wide open
To understand what Y62 phosphorylation does to SHP2, the authors engineered a “phosphomimetic” version (Y62D) that behaves as if Y62 is permanently phosphorylated. Biochemical assays showed that this variant has higher catalytic activity and does not respond normally to activating peptides that would typically “unlock” SHP2 from its closed state. Thermal stability measurements and hydrogen–deuterium exchange experiments revealed that the Y62D protein samples more open, flexible conformations, particularly at the interface between its regulatory and catalytic domains. While a crystal structure of Y62D captured it in a closed form, the dynamic measurements indicated that, in solution, this mutant frequently flips into an open, active-like state, bypassing normal auto-inhibition.
Why current SHP2 drugs struggle in the clinic
Current allosteric SHP2 inhibitors work by stabilizing the closed, inactive shape of SHP2, effectively “gluing” its domains together. The authors showed that the Y62D form of SHP2 is less sensitive to these drugs in purified protein assays. In cancer cell lines where the normal SHP2 gene was replaced with Y62D, growth signaling through the MAPK pathway remained high even when cells were treated with three different clinical-stage SHP2 inhibitors. These cells also maintained phosphorylation of GAB1, a key signaling adaptor that depends on SHP2, highlighting that SHP2 remained functionally active despite drug treatment. Together, the data support a model in which SRC-family kinases phosphorylate SHP2 at Y62, forcing it into an open, active conformation that existing drugs cannot efficiently lock down.
What this means for tomorrow’s cancer therapies
The study identifies SHP2 Y62 phosphorylation as a common, drug-relevant switch that mimics the effect of permanent activating mutations, but without changes to the DNA. This helps explain why cancers driven by growth factor receptors can be intrinsically resistant to current SHP2 inhibitors, even if their SHP2 gene looks normal in sequencing tests. Measuring Y62 phosphorylation may serve as a biomarker to predict which tumors are unlikely to respond to these drugs. More importantly, the work highlights Y62-phosphorylated SHP2 as a distinct structural target and suggests that effective strategies may require either blocking SRC-family kinases upstream, or designing new SHP2 inhibitors that directly recognize and shut down its open, phosphorylated form.
Citation: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Keywords: SHP2, phosphorylation, SRC kinases, MAPK signaling, drug resistance