Clear Sky Science · pl
Miejsce fosforylacji „hotspot” na SHP2 napędza aktywację onkoproteiny i oporność na leki
Dlaczego to ma znaczenie dla przyszłych terapii nowotworów
Wiele nowoczesnych leków przeciwnowotworowych ma na celu wyłączenie nadmiernie aktywnych sygnałów wzrostu wewnątrz komórek nowotworowych. Obiecującym celem był białko o nazwie SHP2, kluczowy przełącznik łączący sygnały z powierzchni komórki z wewnętrznym mechanizmem wzrostu. Mimo to leki zaprojektowane do hamowania SHP2 wykazały wczesnie w badaniach klinicznych niemal brak korzyści. Artykuł ujawnia ukryty „włącznik” na SHP2, który pomaga wyjaśnić, dlaczego te leki często zawodzą, i wskazuje nowe sposoby przechytrzenia opornych guzów.
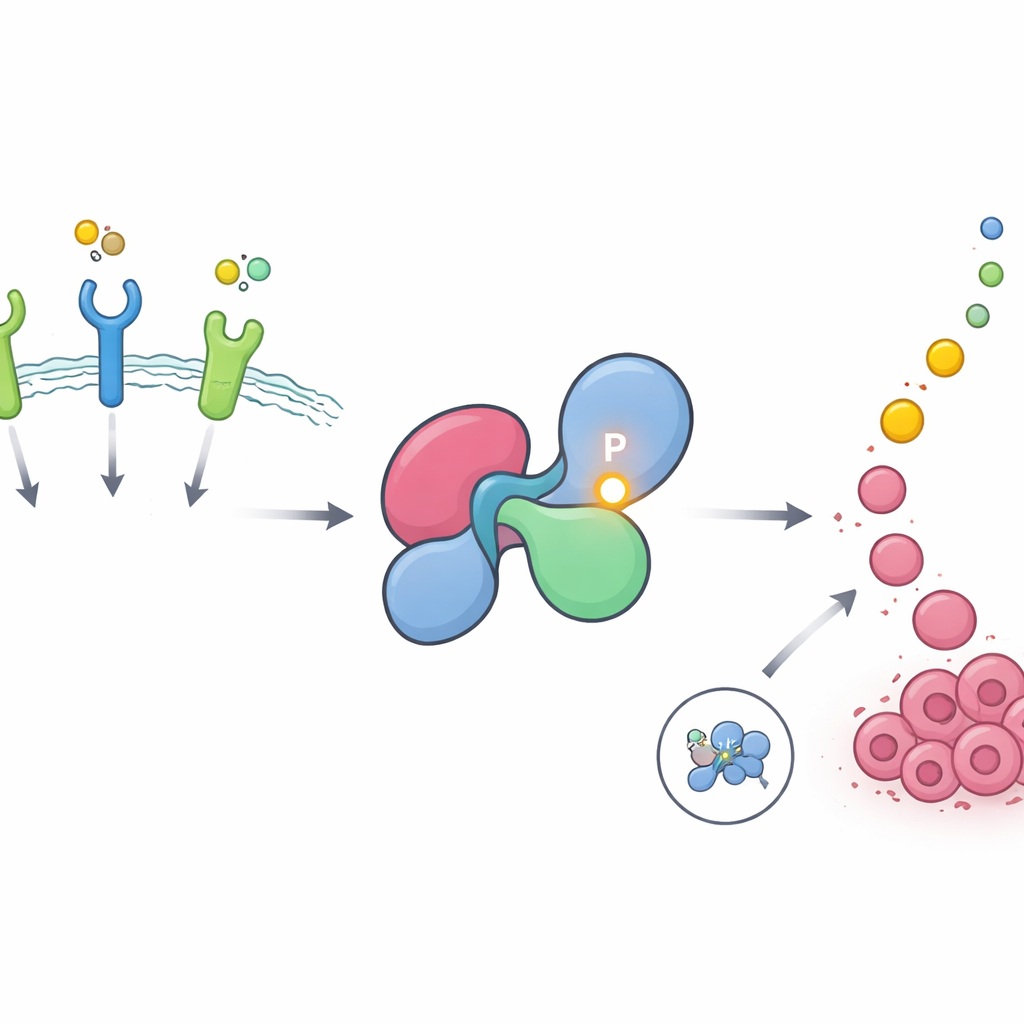
Ukryty punkt wrażliwy na kluczowym przełączniku wzrostu
SHP2 pomaga przekazywać sygnały z receptorów na powierzchni komórki, zwanych receptorowymi kinazami tyrozynowymi, do szlaku RAS–MAPK, głównego motoru wzrostu i podziału komórek. W komórkach zdrowych SHP2 pozostaje przeważnie wyłączony, złożony w zamkniętą konfigurację blokującą jego miejsce aktywne. Krótko się otwiera, gdy receptory są stymulowane, a następnie zamyka ponownie. Autorzy wykorzystali obszerne bazy fosfoproteomiczne, by wyszukać miejsca białkowe często modyfikowane przez dodanie grup fosforanowych — powszechny sposób, w jaki komórki włączają i wyłączają sygnały. Odkryli, że jedno konkretne miejsce na SHP2, tyrozyna 62, jest jednym z najsilniej i najczęściej fosforylowanych miejsc w całym ludzkim proteomie i jest szczególnie wzbogacone w guzach napędzanych receptorami wzrostu takimi jak EGFR i FGFR.
Różne nowotwory, różne poleganie na tym samym przełączniku
Analiza zestawów danych z guzów pacjentów wykazała, że fosforylacja w miejscu Y62 SHP2 jest mocno zwiększona w kilku nowotworach zależnych od receptorów, w tym w niektórych raku płuca, nowotworach dróg żółciowych, guzach głowy i szyi oraz nowotworach mózgu z zaburzeniami EGFR. Co zaskakujące, w niektórych nowotworach płuc fosforylacja Y62 SHP2 była nawet wyższa niż fosforylacja samego receptora. Natomiast guzy napędzane przez zmutowany KRAS, który aktywuje ten sam szlak wzrostu poniżej receptora, wykazywały zmniejszone poziomy fosforylacji Y62 SHP2. Ten wzorzec sugeruje, że nowotwory polegające na sygnałach receptorowych często wzmacniają fosforylację Y62 SHP2 jako element swojej sieci sygnalizacyjnej, podczas gdy te z mutacjami niżej w szlaku nie potrzebują tego konkretnego przełącznika.
Jak inna rodzina kinaz kontroluje ten punkt wrażliwy
Początkowo naturalnym przypuszczeniem było, że same receptory na powierzchni bezpośrednio modyfikują SHP2 w Y62. Jednak gdy zespół zablokował różne receptory przy pomocy celowanych leków, fosforylacja Y62 praktycznie nie zmieniała się. Zamiast tego wyśledzili modyfikację do innej grupy enzymów znanych jako kinazy rodziny SRC. Przy użyciu kombinacji szerokich i selektywnych inhibitorów SRC, komórek z wyciskiem genetycznym SRC, YES1 i FYN oraz oczyszczonych białek w reakcjach in vitro pokazali, że te kinazy bezpośrednio dodają grupy fosforanowe do SHP2 w Y62, a także w dwóch lepiej poznanych miejscach w ogonie SHP2. Umieszcza to kinazy rodziny SRC jako krytycznych pośredników między aktywowanymi receptorami a SHP2, tworząc oś sygnalizacyjną RTK–SRC–SHP2.
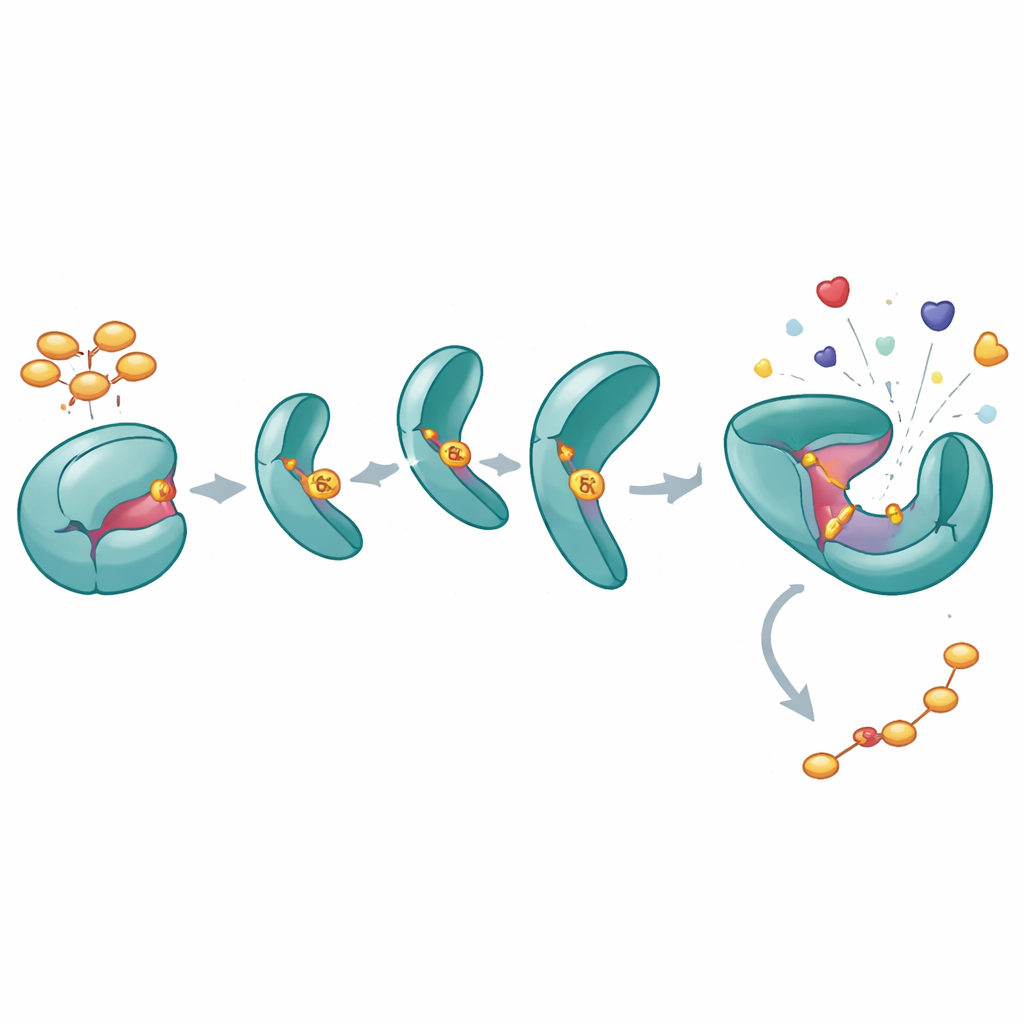
Jak jedna reszta fosforanowa utrzymuje SHP2 szeroko otwarte
Aby zrozumieć, co robi fosforylacja Y62 SHP2, autorzy skonstruowali „fosfomimetyczną” wersję (Y62D), która zachowuje się, jakby Y62 była trwale fosforylowana. Testy biochemiczne wykazały, że wariant ten ma wyższą aktywność katalityczną i nie reaguje normalnie na peptydy aktywujące, które zwykle „odblokowują” SHP2 z jego zamkniętego stanu. Pomiary stabilności termicznej oraz eksperymenty wymiany wodoru na deuter wykazały, że białko Y62D częściej przyjmuje bardziej otwarte, elastyczne konformacje, szczególnie na styku między domenami regulacyjną a katalityczną. Chociaż struktura krystaliczna Y62D utrwaliła je w formie zamkniętej, pomiary dynamiczne wskazały, że w roztworze ta mutacja często przechodzi w stan otwarty przypominający aktywny, omijając normalną auto-inhibicję.
Dlaczego obecne leki na SHP2 mają problemy w klinice
Obecne allosteryczne inhibitory SHP2 działają poprzez stabilizowanie zamkniętej, nieaktywnej formy SHP2, skutecznie „sklejając” jego domeny. Autorzy pokazali, że forma Y62D SHP2 jest mniej wrażliwa na te leki w testach na oczyszczonym białku. W liniach komórkowych nowotworowych, w których normalny gen SHP2 został zastąpiony przez Y62D, sygnalizacja wzrostu przez szlak MAPK pozostawała wysoka nawet po leczeniu trzema różnymi inhibitorami SHP2 będącymi na etapie klinicznym. Komórki te również utrzymywały fosforylację GAB1, kluczowego adaptora sygnalizacyjnego zależnego od SHP2, co podkreśla, że SHP2 pozostawał funkcjonalnie aktywny pomimo leczenia lekiem. Dane te wspierają model, w którym kinazy rodziny SRC fosforylują SHP2 w Y62, wymuszając jego otwartą, aktywną konformację, której istniejące leki nie są w stanie skutecznie zablokować.
Co to oznacza dla jutrzejszych terapii przeciwnowotworowych
Badanie identyfikuje fosforylację Y62 SHP2 jako powszechny, istotny dla leków przełącznik, który naśladuje efekt stałych mutacji aktywujących, ale bez zmian w DNA. To pomaga wyjaśnić, dlaczego nowotwory napędzane przez receptory wzrostu mogą być w sposób pierwotny oporne na obecne inhibitory SHP2, nawet jeśli ich gen SHP2 wygląda normalnie w testach sekwencjonowania. Pomiar fosforylacji Y62 może służyć jako biomarker do przewidywania, które guzy prawdopodobnie nie zareagują na te leki. Co ważniejsze, praca wskazuje na SHP2 z fosforylowanym Y62 jako odrębny cel strukturalny i sugeruje, że skuteczne strategie mogą wymagać albo zablokowania kinaz rodziny SRC powyżej, albo zaprojektowania nowych inhibitorów SHP2, które bezpośrednio rozpoznają i wyłączają jego otwartą, fosforylowaną formę.
Cytowanie: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Słowa kluczowe: SHP2, fosforylacja, kinazy SRC, sygnalizacja MAPK, oporność na leki