Clear Sky Science · fr
Un site de phosphorylation « point chaud » sur SHP2 active l’oncoprotéine et entraîne une résistance aux médicaments
Pourquoi cela compte pour les traitements anticancéreux futurs
De nombreux médicaments anticancéreux modernes visent à couper des signaux de croissance hyperactifs à l’intérieur des cellules tumorales. Une cible prometteuse a été une protéine appelée SHP2, un commutateur clé qui relie les signaux de la surface cellulaire au moteur interne de croissance. Pourtant, les médicaments conçus pour inhiber SHP2 ont montré peu d’efficacité dans les premiers essais cliniques. Cet article révèle un « commutateur » caché sur SHP2 qui aide à expliquer l’échec fréquent de ces traitements et indique de nouvelles voies pour contourner les tumeurs résistantes.
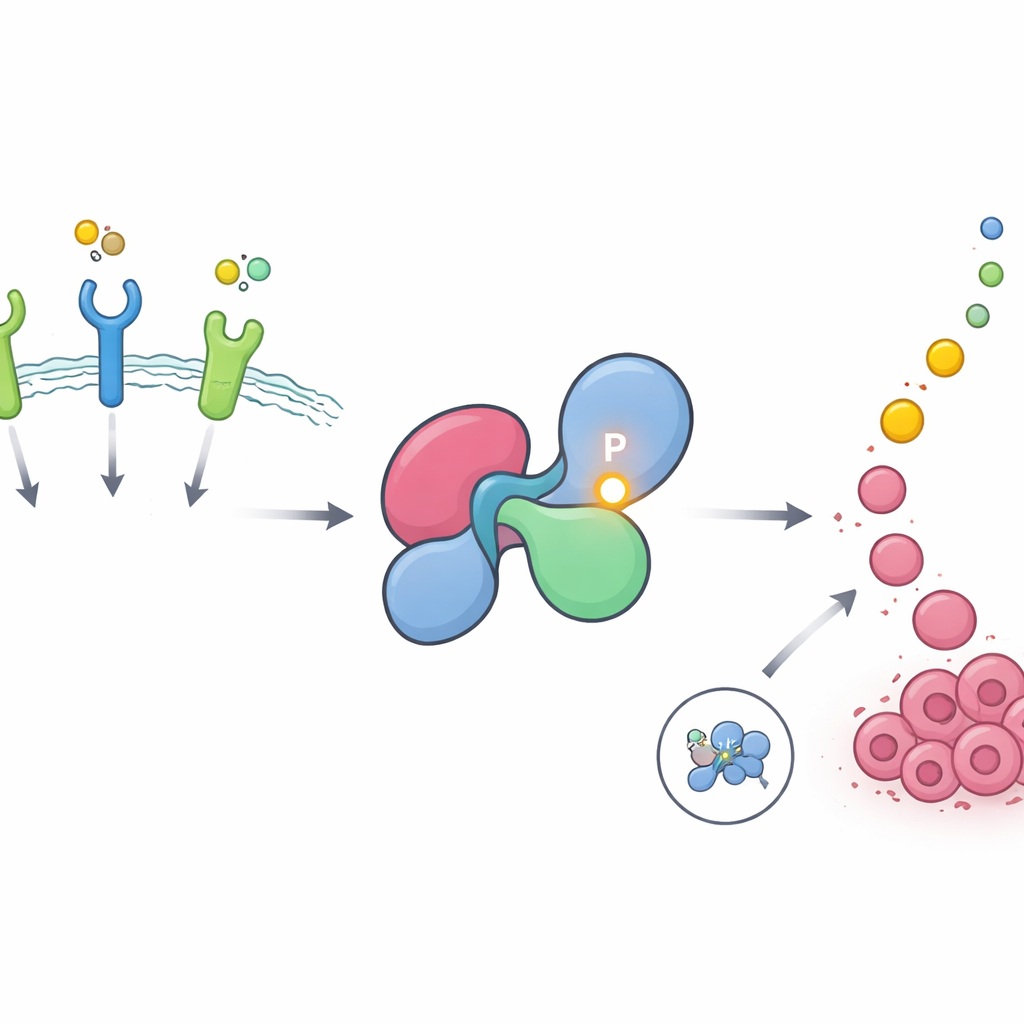
Un point chaud caché sur un commutateur de croissance crucial
SHP2 relaie les messages des récepteurs à la surface cellulaire, appelés récepteurs à activité tyrosine kinase, vers la voie RAS–MAPK, un moteur majeur de la croissance et de la division cellulaire. Dans les cellules saines, SHP2 reste majoritairement inactif, replié dans une conformation fermée qui bloque son site actif. Il s’ouvre brièvement lors de la stimulation des récepteurs, puis se referme. Les auteurs ont utilisé d’importantes bases de données phosphoprotéomiques pour rechercher des sites protéiques fréquemment modifiés par l’addition de groupes phosphate, un moyen courant par lequel les cellules allument ou éteignent des signaux. Ils ont trouvé qu’une position particulière sur SHP2, la tyrosine 62, figure parmi les sites les plus abondamment et récurrentement phosphorylés de tout le protéome humain et est particulièrement enrichie dans les tumeurs pilotées par des récepteurs de facteurs de croissance tels que EGFR et FGFR.
Des cancers différents, une même dépendance au même commutateur
En explorant les jeux de données de tumeurs de patients, les chercheurs ont montré que la phosphorylation sur le site Y62 de SHP2 est fortement augmentée dans plusieurs cancers dépendants de récepteurs, notamment certains cancers du poumon, des voies biliaires, de la tête et du cou, et des tumeurs cérébrales avec altérations d’EGFR. De manière frappante, dans certains cancers du poumon, la phosphorylation de SHP2 en Y62 était même plus élevée que la phosphorylation du récepteur lui‑même. En revanche, les tumeurs pilotées par des mutations de KRAS, qui activent la même voie de croissance en aval, présentaient des niveaux réduits de phosphorylation de SHP2 en Y62. Ce schéma suggère que les cancers dépendants des signaux des récepteurs amplifient souvent Y62 de SHP2 comme partie de leur câblage, tandis que ceux comportant des mutations plus bas dans la voie n’ont pas besoin de ce commutateur particulier.
Comment une autre famille de kinases contrôle ce point chaud
À première vue, il était naturel de suspecter que les récepteurs de surface modifient directement SHP2 en Y62. Cependant, lorsque l’équipe a bloqué divers récepteurs avec des médicaments ciblés, la phosphorylation en Y62 a à peine changé. Ils ont plutôt tracé la modification jusqu’à un autre groupe d’enzymes connues sous le nom de kinases de la famille SRC. En utilisant une combinaison d’inhibiteurs SRC familiaux larges et sélectifs, de cellules knockout dépourvues de SRC, YES1 et FYN, et de protéines purifiées en réaction in vitro, ils ont montré que ces kinases ajoutent directement des groupes phosphate sur SHP2 en Y62, ainsi qu’à deux sites mieux connus sur la queue de SHP2. Cela place les kinases de la famille SRC comme des intermédiaires critiques entre les récepteurs activés et SHP2, créant un axe de signalisation RTK–SRC–SHP2.
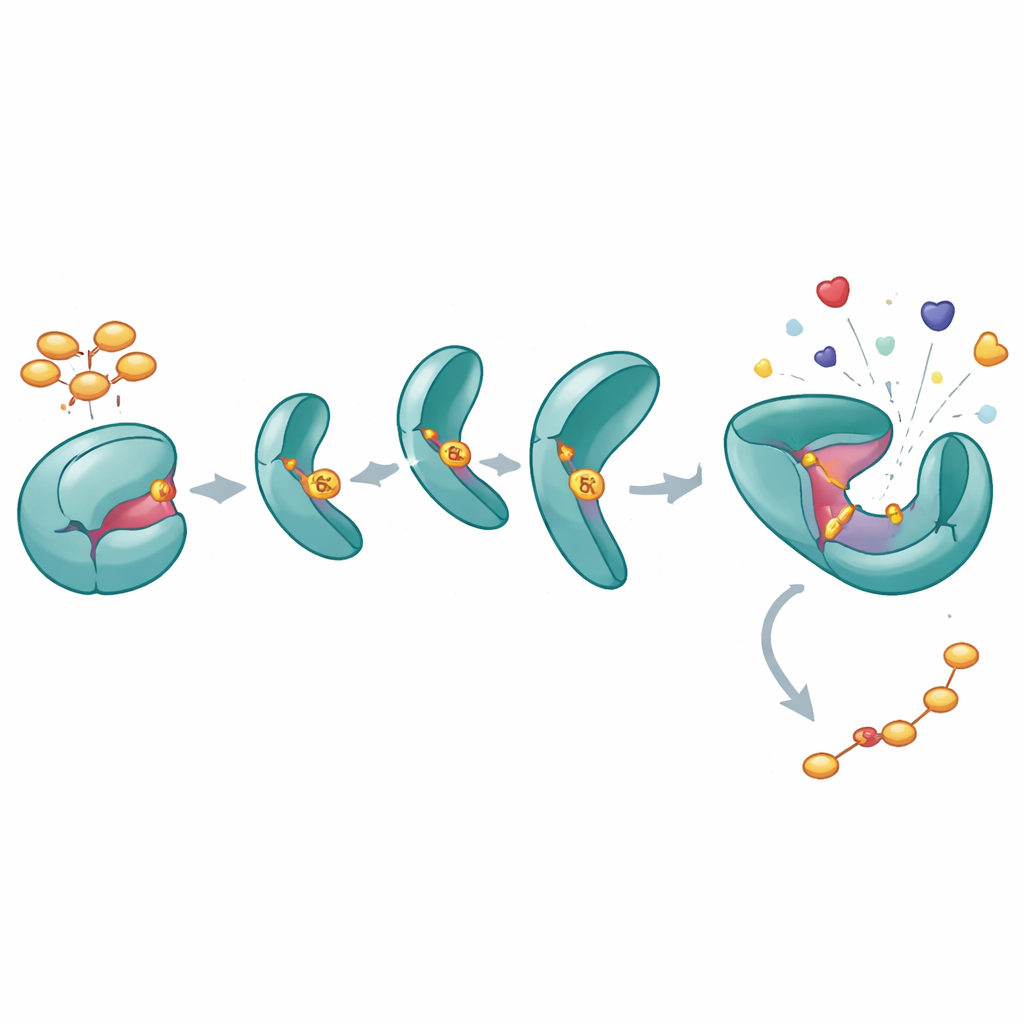
Comment un seul phosphate maintient SHP2 grand ouvert
Pour comprendre l’effet de la phosphorylation en Y62 sur SHP2, les auteurs ont conçu une version « phosphomimétique » (Y62D) qui se comporte comme si Y62 était en permanence phosphorylée. Les essais biochimiques ont montré que cette variante présente une activité catalytique supérieure et ne répond pas normalement aux peptides activateurs qui débloquent habituellement SHP2 de son état fermé. Des mesures de stabilité thermique et des expériences d’échange hydrogène–déutérium ont révélé que la protéine Y62D explore des conformations plus ouvertes et flexibles, en particulier à l’interface entre ses domaines régulateur et catalytique. Alors qu’une structure cristalline de Y62D l’a capturée dans une forme fermée, les mesures dynamiques indiquent qu’en solution ce mutant bascule fréquemment vers un état ouvert, analogue à l’état actif, contournant l’auto‑inhibition normale.
Pourquoi les médicaments actuels ciblant SHP2 peinent en clinique
Les inhibiteurs allostériques actuels de SHP2 agissent en stabilisant la conformation fermée et inactive de SHP2, en « collant » effectivement ses domaines entre eux. Les auteurs ont montré que la forme Y62D de SHP2 est moins sensible à ces médicaments dans des essais sur protéine purifiée. Dans des lignées cellulaires cancéreuses où le gène SHP2 normal a été remplacé par Y62D, la signalisation de croissance via la voie MAPK restait élevée même lorsque les cellules étaient traitées par trois inhibiteurs de SHP2 en phase clinique différents. Ces cellules maintenaient aussi la phosphorylation de GAB1, un adaptateur de signalisation clé dépendant de SHP2, soulignant que SHP2 restait fonctionnellement actif malgré le traitement médicamenteux. Dans l’ensemble, les données soutiennent un modèle dans lequel les kinases de la famille SRC phosphorylent SHP2 en Y62, le forçant en une conformation ouverte et active que les médicaments existants ne parviennent pas efficacement à verrouiller.
Ce que cela signifie pour les thérapies anticancéreuses de demain
L’étude identifie la phosphorylation de SHP2 en Y62 comme un commutateur courant et pertinent pour la thérapeutique, qui mime l’effet de mutations activatrices permanentes, mais sans altération de l’ADN. Cela aide à expliquer pourquoi les cancers pilotés par des récepteurs de facteurs de croissance peuvent être intrinsèquement résistants aux inhibiteurs actuels de SHP2, même si le gène SHP2 paraît normal lors du séquençage. Mesurer la phosphorylation en Y62 pourrait servir de biomarqueur pour prédire quelles tumeurs sont peu susceptibles de répondre à ces médicaments. Plus important encore, le travail met en lumière SHP2 phosphorylé en Y62 comme une cible structurale distincte et suggère que des stratégies efficaces pourraient nécessiter soit de bloquer en amont les kinases de la famille SRC, soit de concevoir de nouveaux inhibiteurs de SHP2 capables de reconnaître et d’inhiber directement sa forme ouverte et phosphorylée.
Citation: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Mots-clés: SHP2, phosphorylation, kinases SRC, signalisation MAPK, résistance aux médicaments