Clear Sky Science · it
Un sito di fosforilazione hotspot su SHP2 guida l’attivazione dell’oncoproteina e la resistenza ai farmaci
Perché questo è importante per i futuri trattamenti contro il cancro
Molti farmaci anticancro moderni puntano a spegnere segnali di crescita iperattivi all’interno delle cellule tumorali. Un bersaglio promettente è stato una proteina chiamata SHP2, un interruttore chiave che collega i segnali della superficie cellulare al motore interno della crescita. Tuttavia, i farmaci progettati per inibire SHP2 hanno mostrato quasi nessun beneficio nelle prime sperimentazioni cliniche. Questo studio individua un “interruttore acceso” nascosto su SHP2 che aiuta a spiegare perché questi farmaci spesso falliscono e indica nuovi modi per aggirare i tumori resistenti.
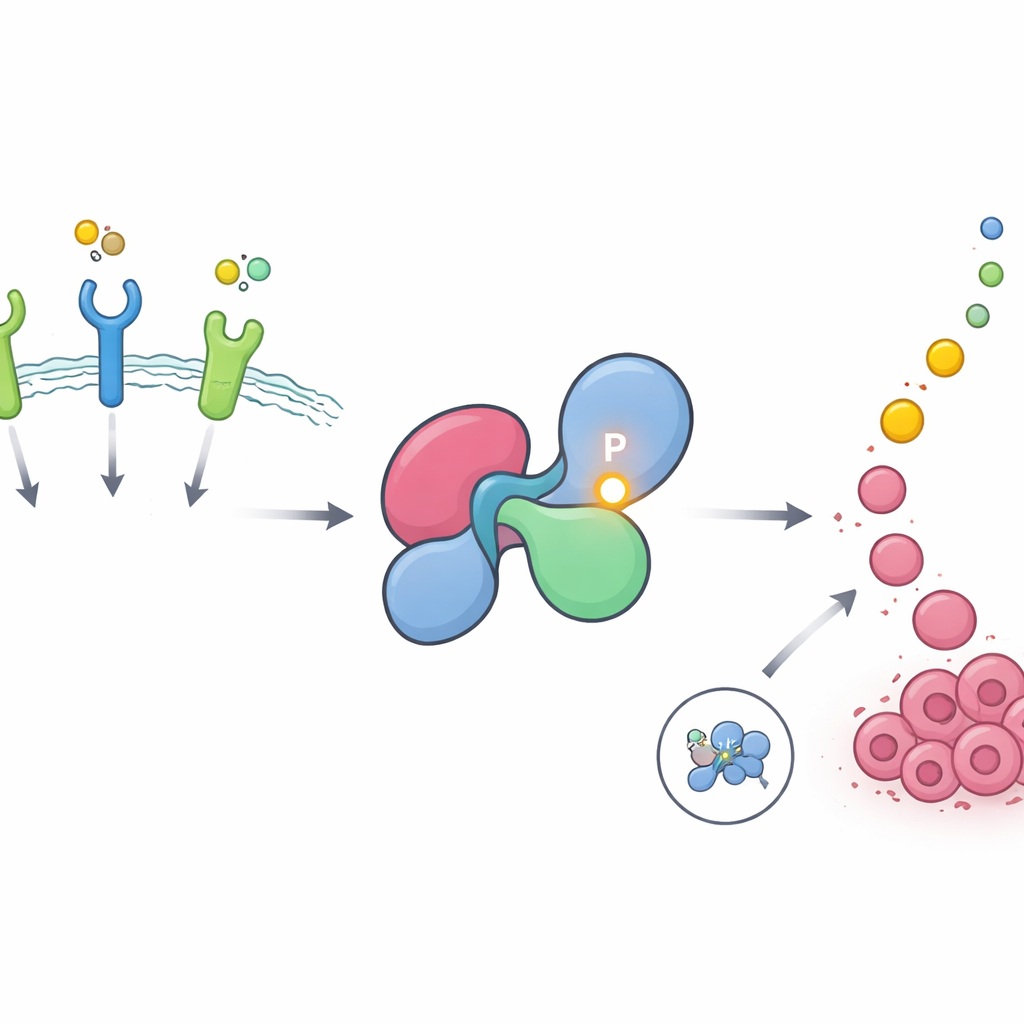
Un punto caldo nascosto su un interruttore di crescita cruciale
SHP2 contribuisce a trasmettere messaggi dai recettori sulla superficie cellulare, detti recettori tirosin-chinasici, alla via RAS–MAPK, un motore principale della crescita e della divisione cellulare. Nelle cellule sane, SHP2 rimane per lo più spento, ripiegato in una conformazione chiusa che blocca il suo sito attivo. Si apre brevemente quando i recettori vengono stimolati, quindi si richiude. Gli autori hanno usato grandi database di fosfoproteomica per cercare siti proteici frequentemente modificati dall’aggiunta di gruppi fosfato, un modo comune con cui le cellule accendono o spengono i segnali. Hanno trovato che una particolare posizione su SHP2, chiamata tirosina 62, è tra i siti più intensamente e ricorrentemente fosforilati nell’intero proteoma umano ed è particolarmente arricchita nei tumori guidati da recettori di fattori di crescita come EGFR e FGFR.
Cancro diverso, dipendenza diversa dallo stesso interruttore
Analizzando i dataset di tumori dei pazienti, i ricercatori hanno mostrato che la fosforilazione nel sito Y62 di SHP2 è fortemente aumentata in diversi tumori guidati dai recettori, inclusi alcuni tumori polmonari, tumori delle vie biliari, neoplasie della testa e del collo e tumori cerebrali con alterazioni di EGFR. Colpisce che, in alcuni tumori polmonari, la fosforilazione di SHP2 Y62 risultava ancora più elevata della fosforilazione sul recettore stesso. Al contrario, i tumori guidati da KRAS mutato, che attiva la stessa via di crescita a valle, mostravano livelli ridotti di fosforilazione di SHP2 Y62. Questo schema suggerisce che i tumori che dipendono dai segnali dei recettori spesso potenziano Y62 di SHP2 come parte del loro cablaggio, mentre quelli con mutazioni più a valle non necessitano di questo particolare interruttore.
Come un’altra famiglia di chinasi controlla questo punto caldo
A prima vista, era naturale sospettare che i recettori di superficie modificassero direttamente SHP2 in Y62. Tuttavia, quando il gruppo ha bloccato vari recettori con farmaci mirati, la fosforilazione di Y62 è cambiata di poco. Invece, hanno ricondotto la modifica a un diverso gruppo di enzimi noti come chinasi della famiglia SRC. Usando una combinazione di inibitori della famiglia SRC ampi e selettivi, linee cellulari con knockout genetico di SRC, YES1 e FYN, e proteine purificate in reazioni in vitro, hanno dimostrato che queste chinasi aggiungono direttamente gruppi fosfato a SHP2 in Y62, e anche in due siti più noti sulla coda di SHP2. Questo colloca le chinasi della famiglia SRC come mediatori critici tra i recettori attivati e SHP2, creando un asse di segnalazione RTK–SRC–SHP2.
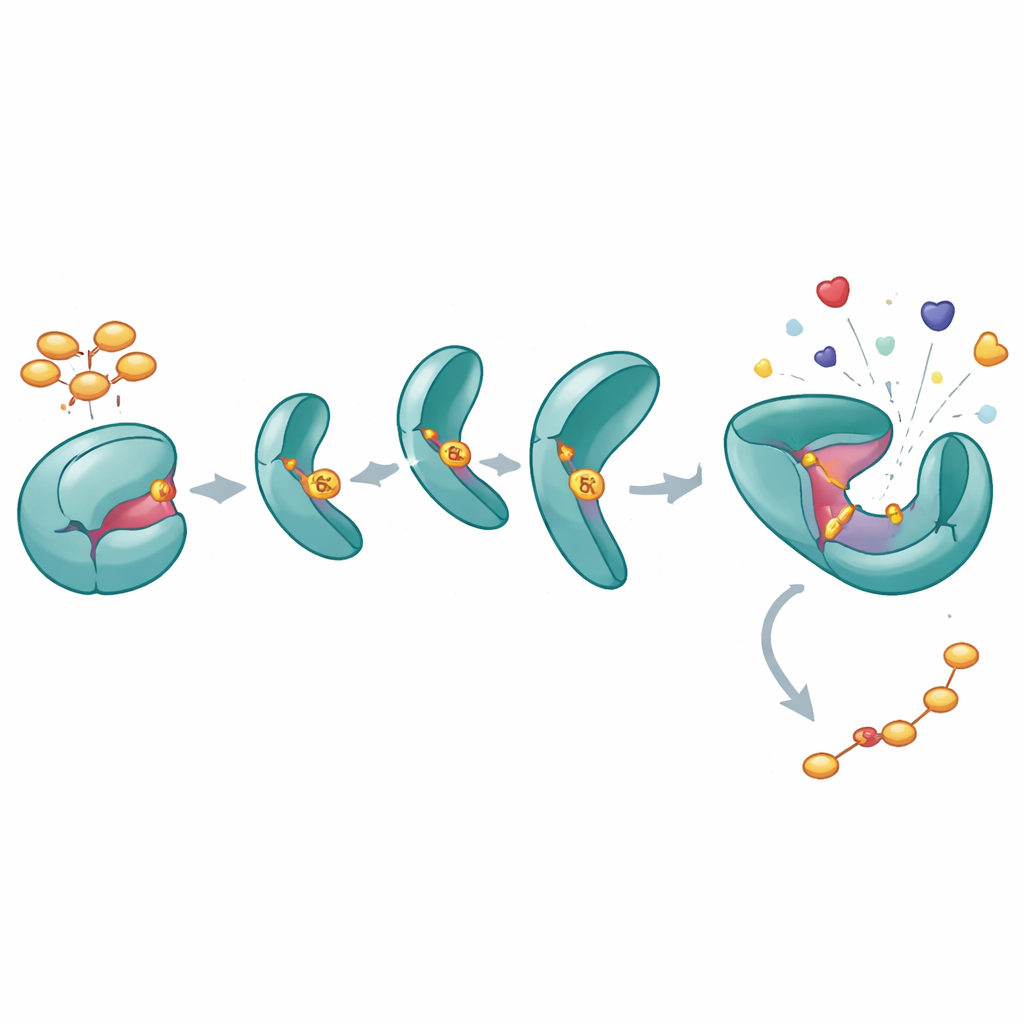
Come un singolo fosfato tiene SHP2 spalancato
Per capire cosa provoca la fosforilazione di Y62 su SHP2, gli autori hanno costruito una versione “fosfomimetica” (Y62D) che si comporta come se Y62 fosse permanentemente fosforilata. Saggi biochimici hanno mostrato che questa variante ha un’attività catalitica più elevata e non risponde normalmente ai peptidi attivatori che tipicamente “sbloccano” SHP2 dalla sua forma chiusa. Misure di stabilità termica e esperimenti di scambio idrogeno-deuterio hanno rivelato che la proteina Y62D esplora conformazioni più aperte e flessibili, in particolare all’interfaccia tra i domini regolatori e catalitici. Mentre una struttura cristallina di Y62D la catturava in una forma chiusa, le misure dinamiche indicano che, in soluzione, questo mutante passa frequentemente in uno stato aperto, simile all’attivo, superando l’auto-inibizione normale.
Perché gli attuali farmaci per SHP2 faticano in clinica
Gli inibitori allosterici di SHP2 attuali funzionano stabilizzando la forma chiusa e inattiva di SHP2, «incollando» efficacemente i suoi domini. Gli autori hanno dimostrato che la forma Y62D di SHP2 è meno sensibile a questi farmaci in saggi su proteine purificate. In linee cellulari tumorali in cui il gene SHP2 normale è stato sostituito con Y62D, la segnalazione di crescita attraverso la via MAPK rimaneva elevata anche quando le cellule venivano trattate con tre diversi inibitori clinici di SHP2. Queste cellule mantenevano inoltre la fosforilazione di GAB1, un adattatore di segnalazione chiave dipendente da SHP2, sottolineando che SHP2 rimaneva funzionalmente attivo nonostante il trattamento farmacologico. Nel complesso, i dati supportano un modello in cui le chinasi della famiglia SRC fosforilano SHP2 in Y62, costringendolo in una conformazione aperta e attiva che gli inibitori esistenti non riescono a bloccare efficacemente.
Cosa significa questo per le terapie oncologiche di domani
Lo studio identifica la fosforilazione di SHP2 in Y62 come un interruttore comune e rilevante per i farmaci che imita l’effetto di mutazioni attivanti permanenti, ma senza alterazioni del DNA. Questo aiuta a spiegare perché i tumori guidati da recettori di fattori di crescita possono essere intrinsecamente resistenti agli inibitori di SHP2 attuali, anche se il gene SHP2 appare normale nei test di sequenziamento. Misurare la fosforilazione di Y62 potrebbe servire come biomarcatore per prevedere quali tumori probabilmente non risponderanno a questi farmaci. Più importante, il lavoro evidenzia SHP2 fosforilato in Y62 come un bersaglio strutturale distinto e suggerisce che strategie efficaci potrebbero richiedere o il blocco a monte delle chinasi della famiglia SRC, o la progettazione di nuovi inibitori di SHP2 che riconoscano direttamente e spengano la sua forma aperta e fosforilata.
Citazione: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Parole chiave: SHP2, fosforilazione, chinasi SRC, segnalazione MAPK, resistenza ai farmaci