Clear Sky Science · es
Un sitio de fosforilación prioritario en SHP2 impulsa la activación del oncoproteína y la resistencia a fármacos
Por qué esto importa para los tratamientos oncológicos futuros
Muchos fármacos oncológicos modernos intentan desactivar señales de crecimiento hiperactivas dentro de las células tumorales. Un objetivo prometedor ha sido una proteína llamada SHP2, un interruptor clave que conecta las señales en la superficie celular con el motor de crecimiento interno de la célula. Sin embargo, los fármacos diseñados para inhibir SHP2 han mostrado casi ningún beneficio en ensayos clínicos iniciales. Este artículo descubre un “interruptor encendido” oculto en SHP2 que ayuda a explicar por qué estos fármacos a menudo fallan y apunta a nuevas maneras de burlar tumores resistentes.
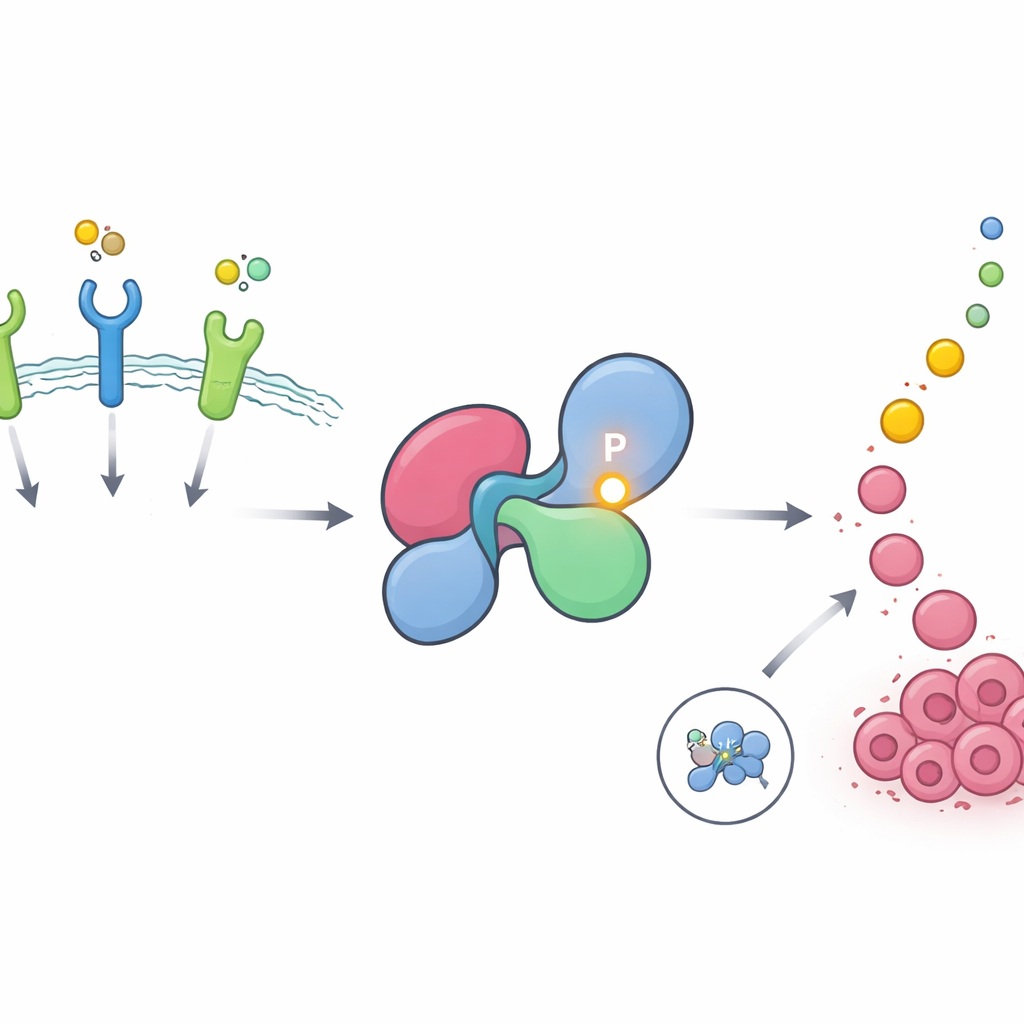
Un punto caliente oculto en un interruptor de crecimiento crucial
SHP2 ayuda a transmitir mensajes desde receptores en la superficie celular, llamados receptores tirosina quinasa, hasta la vía RAS–MAPK, un impulsor principal del crecimiento y la división celular. En células sanas, SHP2 permanece en gran medida apagado, plegado en una conformación cerrada que bloquea su propio sitio activo. Se abre brevemente cuando los receptores se estimulan y luego se cierra de nuevo. Los autores usaron grandes bases de datos fosfoproteómicas para buscar sitios proteicos que sean frecuentemente modificados por la adición de grupos fosfato, una forma habitual en que las células activan o desactivan señales. Encontraron que una posición particular en SHP2, denominada tirosina 62, está entre los sitios más fuertemente y recurrentemente fosforilados en todo el proteoma humano y está especialmente enriquecida en tumores impulsados por receptores de factores de crecimiento como EGFR y FGFR.
Diferentes cánceres, distinta dependencia del mismo interruptor
Al analizar conjuntos de datos de tumores de pacientes, los investigadores mostraron que la fosforilación en el sitio Y62 de SHP2 aumenta de forma marcada en varios cánceres impulsados por receptores, incluidos ciertos cánceres de pulmón, del conducto biliar, de cabeza y cuello y tumores cerebrales con alteraciones en EGFR. Sorprendentemente, en algunos cánceres de pulmón, la fosforilación de Y62 en SHP2 estaba incluso más elevada que la fosforilación del propio receptor. En contraste, los tumores impulsados por KRAS mutante, que activa la misma vía de crecimiento más abajo, mostraron niveles reducidos de fosforilación de SHP2 Y62. Este patrón sugiere que los cánceres que dependen de señales receptoras a menudo aumentan la fosforilación de SHP2 Y62 como parte de su cableado, mientras que aquellos con mutaciones más abajo en la vía no necesitan este interruptor en particular.
Cómo otra familia de quinasas controla este punto caliente
A primera vista, era natural sospechar que los receptores de superficie modificaban directamente SHP2 en Y62. Sin embargo, cuando el equipo bloqueó varios receptores con fármacos dirigidos, la fosforilación de Y62 apenas cambió. En su lugar, rastrearon la modificación hasta un grupo diferente de enzimas conocidas como quinasas de la familia SRC. Usando una combinación de inhibidores de la familia SRC de amplio y selectivo espectro, células con eliminación genética de SRC, YES1 y FYN, y proteínas purificadas en reacciones in vitro, demostraron que estas quinasas añaden directamente grupos fosfato a SHP2 en Y62, y también en dos sitios más conocidos en la cola de SHP2. Esto coloca a las quinasas de la familia SRC como mediadores críticos que se sitúan entre receptores activados y SHP2, creando un eje de señalización RTK–SRC–SHP2.
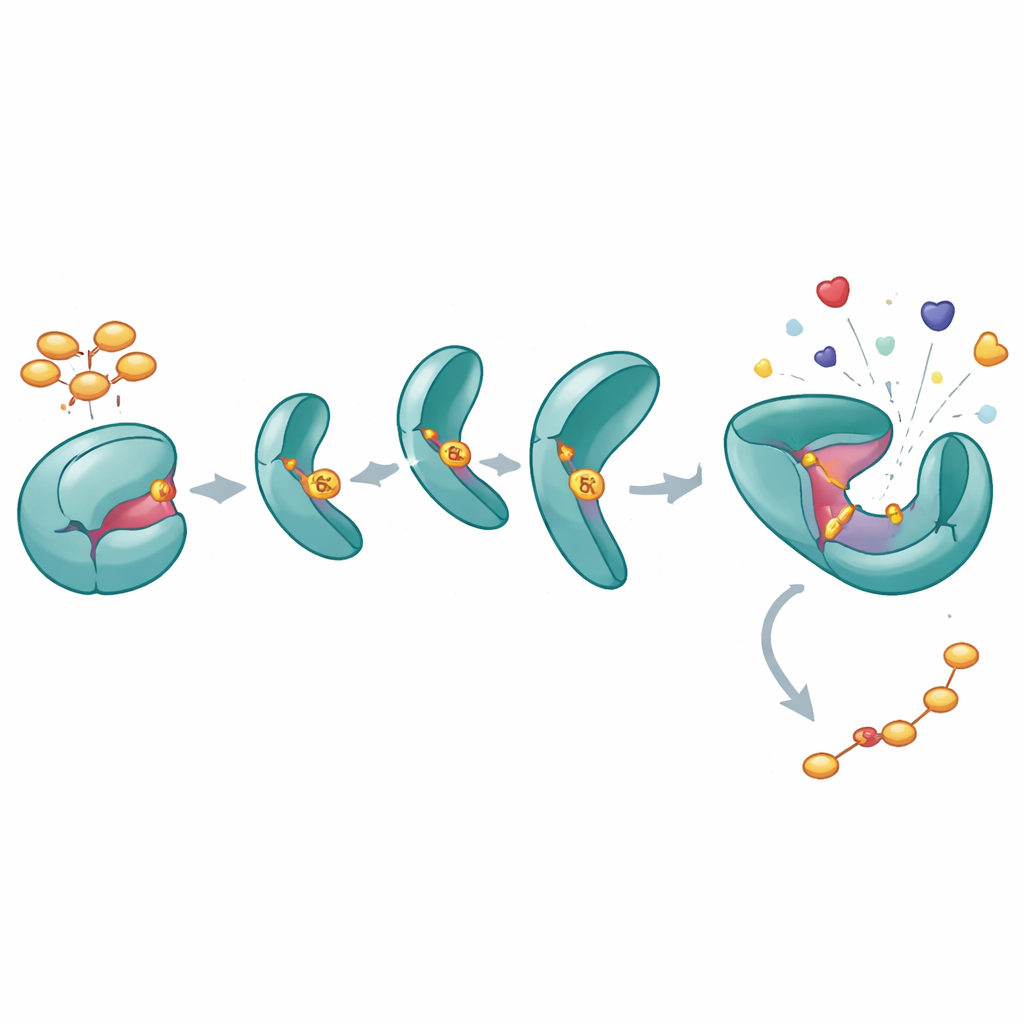
Cómo un fosfato mantiene a SHP2 completamente abierto
Para entender qué hace la fosforilación en Y62 a SHP2, los autores diseñaron una versión “fosfomimética” (Y62D) que se comporta como si Y62 estuviera permanentemente fosforilada. Ensayos bioquímicos mostraron que esta variante tiene mayor actividad catalítica y no responde normalmente a los péptidos activadores que típicamente “desbloquearían” a SHP2 de su estado cerrado. Mediciones de estabilidad térmica y experimentos de intercambio hidrógeno–deuterio revelaron que la proteína Y62D explora conformaciones más abiertas y flexibles, particularmente en la interfaz entre sus dominios regulador y catalítico. Aunque una estructura cristalina de Y62D la capturó en una forma cerrada, las mediciones dinámicas indicaron que, en solución, este mutante frecuentemente cambia a un estado abierto, similar al activo, eludiendo la auto-inhibición normal.
Por qué los fármacos actuales contra SHP2 tienen dificultades en la clínica
Los inhibidores alostéricos actuales de SHP2 actúan estabilizando la forma cerrada e inactiva de SHP2, efectivamente “pegando” sus dominios entre sí. Los autores mostraron que la forma Y62D de SHP2 es menos sensible a estos fármacos en ensayos con proteínas purificadas. En líneas celulares cancerosas donde el gen SHP2 normal fue reemplazado por Y62D, la señalización de crecimiento a través de la vía MAPK permaneció alta incluso cuando las células se trataron con tres distintos inhibidores de SHP2 en fase clínica. Estas células también mantuvieron la fosforilación de GAB1, un adaptador de señalización clave que depende de SHP2, lo que subraya que SHP2 seguía funcionalmente activo a pesar del tratamiento farmacológico. En conjunto, los datos apoyan un modelo en el que las quinasas de la familia SRC fosforilan SHP2 en Y62, forzándolo a una conformación abierta y activa que los fármacos existentes no pueden bloquear de manera eficiente.
Qué significa esto para las terapias oncológicas del mañana
El estudio identifica la fosforilación de SHP2 Y62 como un interruptor común y relevante para fármacos que imita el efecto de mutaciones activadoras permanentes, pero sin cambios en el ADN. Esto ayuda a explicar por qué los cánceres impulsados por receptores de factores de crecimiento pueden ser intrínsecamente resistentes a los inhibidores actuales de SHP2, incluso si su gen SHP2 parece normal en pruebas de secuenciación. Medir la fosforilación de Y62 podría servir como biomarcador para predecir qué tumores probablemente no responderán a estos fármacos. Más importante aún, el trabajo destaca a SHP2 fosforilado en Y62 como un objetivo estructural distinto y sugiere que las estrategias efectivas podrían requerir bloquear las quinasas de la familia SRC aguas arriba, o diseñar nuevos inhibidores de SHP2 que reconozcan directamente y clausuren su forma abierta y fosforilada.
Cita: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Palabras clave: SHP2, fosforilación, quinasas SRC, señalización MAPK, resistencia a fármacos