Clear Sky Science · pt
Um sítio de fosforilação hotspot em SHP2 impulsiona ativação de oncoproteína e resistência a medicamentos
Por que isso importa para tratamentos de câncer no futuro
Muitos medicamentos modernos contra o câncer buscam desligar sinais de crescimento hiperativos dentro das células tumorais. Um alvo promissor tem sido uma proteína chamada SHP2, um interruptor chave que conecta sinais na superfície celular ao motor interno de crescimento da célula. Ainda assim, fármacos projetados para inibir o SHP2 mostraram quase nenhum benefício em ensaios clínicos iniciais. Este artigo revela um “botão liga” oculto no SHP2 que ajuda a explicar por que esses medicamentos frequentemente falham e aponta novas formas de contornar tumores resistentes.
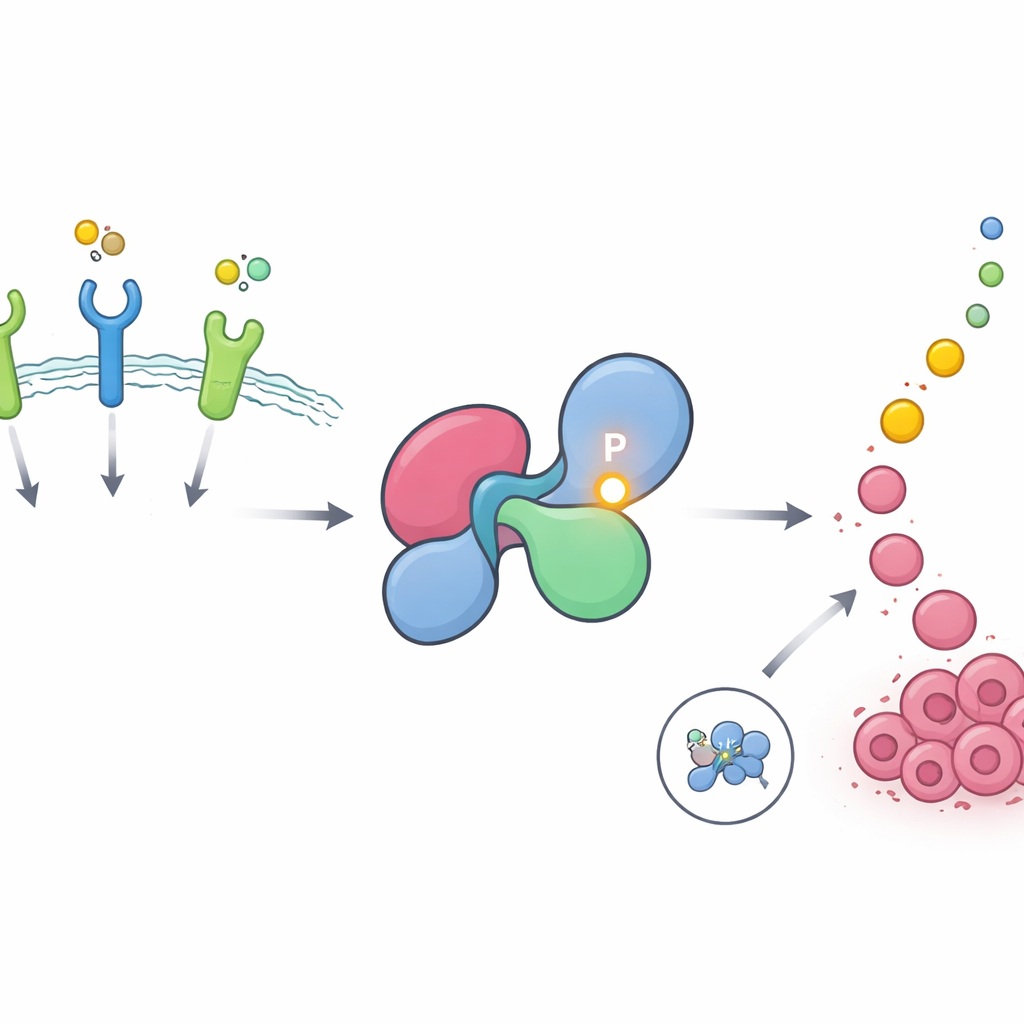
Um ponto crítico oculto em um interruptor de crescimento crucial
O SHP2 ajuda a transmitir mensagens de receptores na superfície celular, chamados receptores tirosina quinase, para a via RAS–MAPK, um motor importante do crescimento e divisão celular. Em células saudáveis, o SHP2 fica majoritariamente desligado, dobrado em uma forma fechada que bloqueia seu próprio sítio ativo. Ele se abre brevemente quando os receptores são estimulados e depois se fecha novamente. Os autores usaram grandes bancos de dados fosfoproteômicos para procurar sítios proteicos frequentemente modificados pela adição de grupos fosfato, uma forma comum de as células ligarem e desligarem sinais. Eles descobriram que uma posição específica no SHP2, chamada tirosina 62, está entre os sítios mais intensamente e recorrentemente fosforilados em todo o proteoma humano e está especialmente enriquecida em tumores dirigidos por receptores de fatores de crescimento como EGFR e FGFR.
Diferentes cânceres, diferente dependência do mesmo interruptor
Ao vasculhar conjuntos de dados de tumores de pacientes, os pesquisadores mostraram que a fosforilação no sítio Y62 do SHP2 aumenta fortemente em vários cânceres impulsionados por receptores, incluindo certos cânceres de pulmão, cânceres das vias biliares, tumores de cabeça e pescoço e tumores cerebrais com alterações em EGFR. De forma marcante, em alguns cânceres de pulmão, a fosforilação de Y62 no SHP2 estava ainda mais elevada do que a fosforilação no próprio receptor. Em contraste, tumores impulsionados por KRAS mutante, que ativam a mesma via de crescimento mais adiante, mostraram níveis reduzidos de fosforilação de SHP2 Y62. Esse padrão sugere que cânceres que dependem de sinais dos receptores frequentemente aumentam a fosforilação de SHP2 Y62 como parte de sua fiação, enquanto aqueles com mutações mais abaixo na via não precisam desse interruptor em particular.
Como outra família de quinases controla esse ponto crítico
À primeira vista, parecia natural suspeitar que os próprios receptores de superfície modificassem diretamente o SHP2 em Y62. No entanto, quando a equipe bloqueou vários receptores com medicamentos direcionados, a fosforilação em Y62 mal se alterou. Em vez disso, eles rastrearam a modificação até um grupo diferente de enzimas conhecidas como quinases da família SRC. Usando uma combinação de inibidores amplos e seletivos da família SRC, células com nocaute genético para SRC, YES1 e FYN, e proteínas purificadas em reações de tubo de ensaio, demonstraram que essas quinases adicionam diretamente grupos fosfato ao SHP2 em Y62, e também em dois sítios mais conhecidos na cauda do SHP2. Isso posiciona as quinases da família SRC como intermediárias críticas entre receptores ativados e o SHP2, criando um eixo de sinalização RTK–SRC–SHP2.
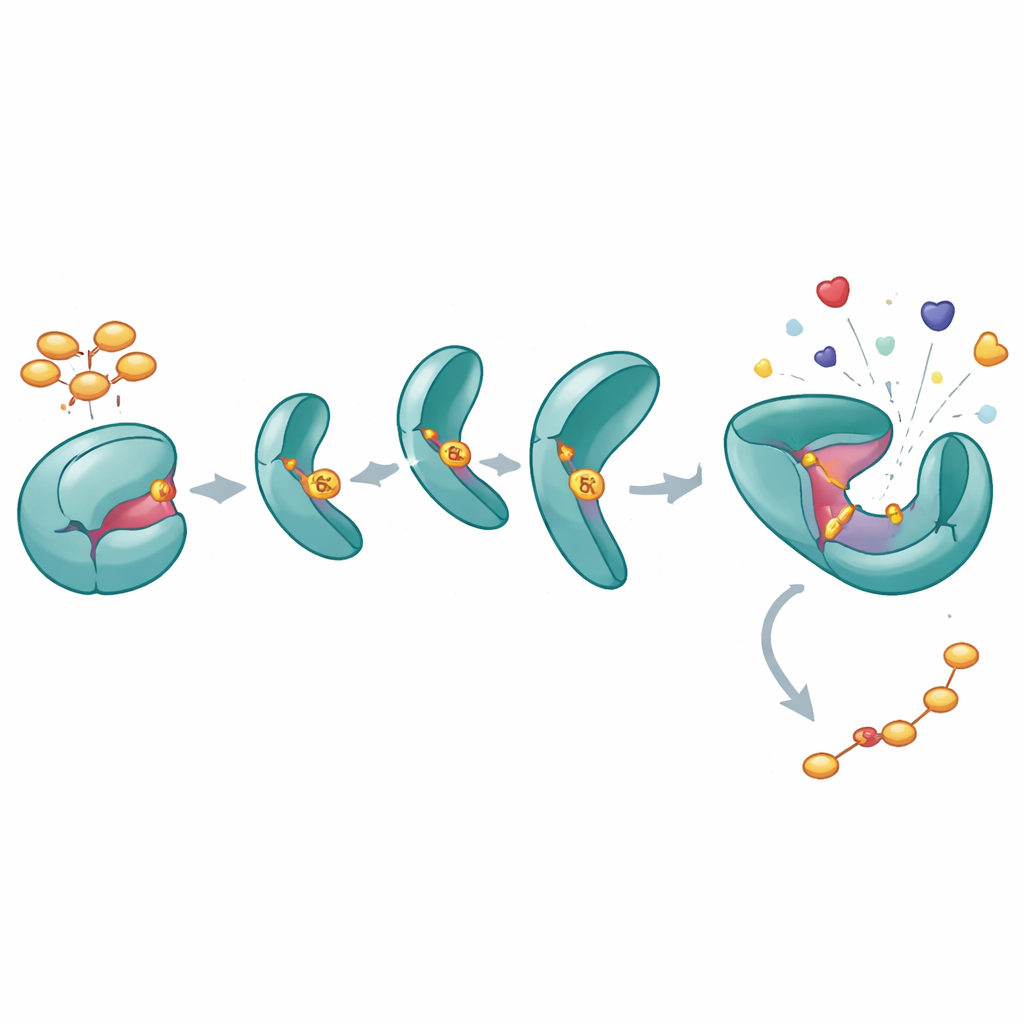
Como um único fosfato mantém o SHP2 bem aberto
Para entender o que a fosforilação de Y62 faz ao SHP2, os autores criaram uma versão “fosfomimética” (Y62D) que se comporta como se Y62 estivesse permanentemente fosforilado. Ensaios bioquímicos mostraram que essa variante tem maior atividade catalítica e não responde normalmente a peptídeos ativadores que tipicamente “destravariam” o SHP2 de seu estado fechado. Medidas de estabilidade térmica e experimentos de troca hidrogênio–deutério revelaram que a proteína Y62D acessa conformações mais abertas e flexíveis, particularmente na interface entre seus domínios regulatório e catalítico. Embora uma estrutura cristalina de Y62D a tenha capturado em uma forma fechada, as medidas dinâmicas indicaram que, em solução, esse mutante frequentemente alterna para um estado aberto, semelhante ao ativo, contornando a inibição automática normal.
Por que os medicamentos atuais contra SHP2 têm dificuldade na clínica
Os inibidores alostéricos atuais do SHP2 atuam estabilizando a forma fechada e inativa do SHP2, efetivamente “colando” seus domínios juntos. Os autores mostraram que a forma Y62D do SHP2 é menos sensível a esses medicamentos em ensaios com proteína purificada. Em linhagens celulares cancerosas em que o gene SHP2 normal foi substituído por Y62D, a sinalização de crescimento via via MAPK permaneceu alta mesmo quando as células foram tratadas com três diferentes inibidores de SHP2 em estágio clínico. Essas células também mantiveram a fosforilação de GAB1, um adaptador de sinalização chave que depende do SHP2, destacando que o SHP2 permaneceu funcionalmente ativo apesar do tratamento medicamentoso. Em conjunto, os dados sustentam um modelo no qual quinases da família SRC fosforilam o SHP2 em Y62, forçando-o a uma conformação aberta e ativa que os medicamentos existentes não conseguem travar eficientemente.
O que isso significa para as terapias contra o câncer de amanhã
O estudo identifica a fosforilação de SHP2 Y62 como um interruptor comum e relevante para medicamentos que mimetiza o efeito de mutações ativadoras permanentes, mas sem alterações no DNA. Isso ajuda a explicar por que cânceres dirigidos por receptores de fatores de crescimento podem ser intrinsecamente resistentes aos inibidores atuais de SHP2, mesmo quando o gene SHP2 parece normal em testes de sequenciamento. Medir a fosforilação de Y62 pode servir como biomarcador para prever quais tumores provavelmente não responderão a esses medicamentos. Mais importante, o trabalho destaca o SHP2 fosforilado em Y62 como um alvo estrutural distinto e sugere que estratégias eficazes podem exigir ou bloquear as quinases da família SRC a montante, ou projetar novos inibidores de SHP2 que reconheçam e desliguem diretamente sua forma aberta e fosforilada.
Citação: Karunaraj, P., Scheele, R., Wells, M.L. et al. A hotspot phosphorylation site on SHP2 drives oncoprotein activation and drug resistance. Nat Commun 17, 3383 (2026). https://doi.org/10.1038/s41467-026-70060-8
Palavras-chave: SHP2, fosforilação, quinasas SRC, sinalização MAPK, resistência a medicamentos