Clear Sky Science · ru
BACH2 связывает сигнализацию β1-адренорецептора/β-аррестина1 с MIAT, ингибируя активацию кардиальных фибробластов и апоптоз кардиомиоцитов
Почему это исследование сердца важно
Инфаркты и хроническая сердечная недостаточность по-прежнему остаются одними из ведущих причин смерти, отчасти потому, что поврежденная сердечная мышца замещается рубцовой тканью, а жизненно важные клетки сердца погибают. Это исследование обнаруживает ранее неизвестный предохранительный механизм внутри сердечных клеток, который включается при действии широко применяемого бета‑блокатора и, в свою очередь, отключает вредный РНК‑сигнал, связанный с образованием рубца и гибелью клеток. Понимание этого внутреннего защитного пути может указать путь к более точным терапиям, которые помогут поврежденным сердцам дольше и лучше сохранять функцию.
Вредное «сообщение» в сердцах с недостаточностью
Когда часть сердца лишается крови, многие работающие мышечные клетки (кардиомиоциты) погибают и замещаются жестким рубцом, который продуцируют поддерживающие клетки — фибробласты. Ранее в исследованиях была описана длинная некодирующая РНК под названием MIAT, действующая как токсичное «сообщение»: она усиливает экспрессию генов, способствующих фиброзу и запрограммированной гибели клеток. Уровни MIAT повышаются при нескольких заболеваниях сердца у людей и в ряде животных моделей повреждения сердца, а блокирование MIAT у мышей улучшает функцию сердца после инфаркта. Однако до сих пор было неизвестно, что регулирует саму MIAT и как стандартные сердечные препараты на неё влияют.
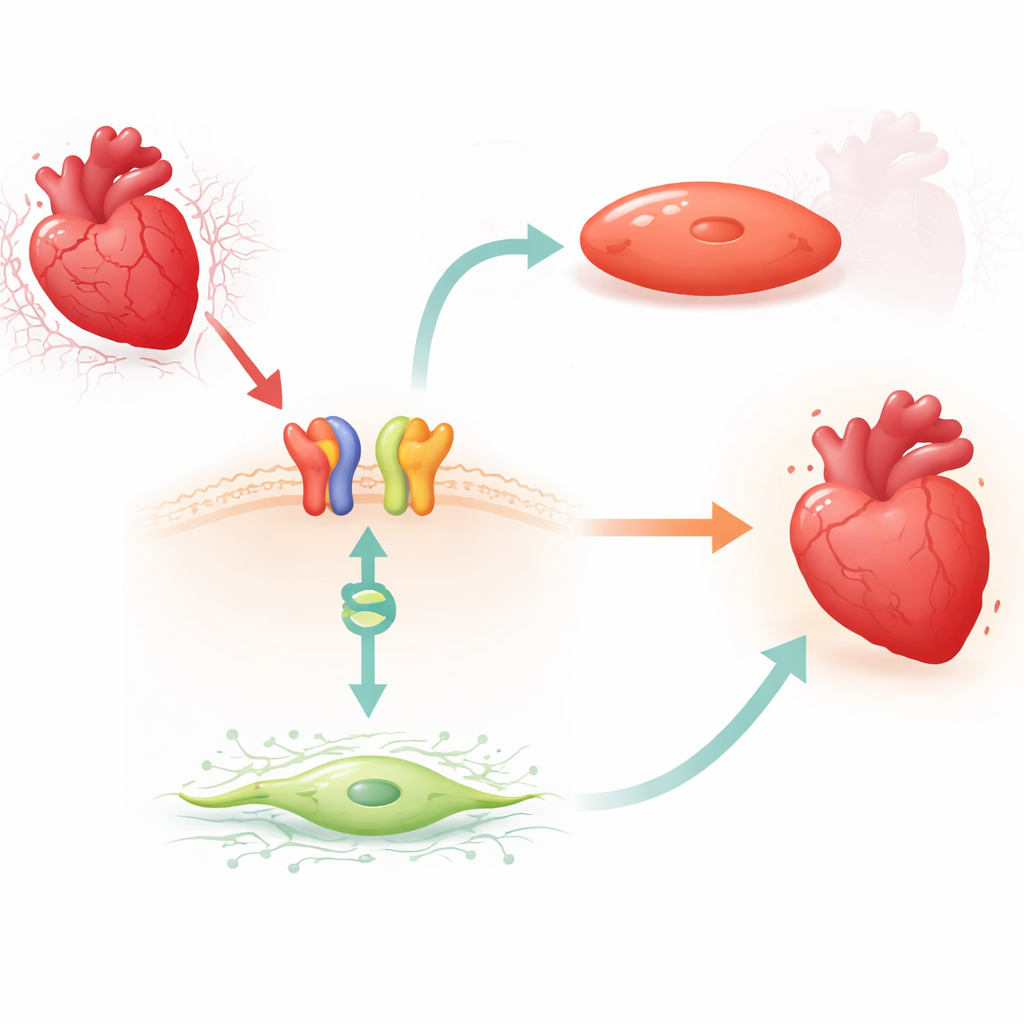
Связь от поверхности клетки к переключателю гена, активируемая лекарством
Исследователи сосредоточились на карведилоле, широко используемом бета‑блокаторе, который может запускать защитный сигнальный путь через β1‑адренорецептор и адапторный белок β‑аррестин1. У мышей они обнаружили, что короткое лечение карведилолом последовательно снижало уровни MIAT в левом желудочке, но только при наличии β1-рецепторов и β‑аррестин1. Похожее снижение MIAT наблюдалось в культивируемых взрослых человеческих кардиальных фибробластах и в человеческих и грызуньих кардиомиоцитах, подвергнутых лабораторной модели гипоксии и реперфузии. В этих системах стресс повышал MIAT, тогда как карведилол обращал это повышение, что указывает на то, что препарат задействует естеительный тормоз вредной РНК.
Защитный белок, который подавляет вредную РНК
Углубившись, команда искала ДНК‑связывающий белок, который мог бы взаимодействовать с β‑аррестин1 в ядре клетки. Они сосредоточились на BACH2 — факторе транскрипции, ранее связанном с защитой от других форм повреждения сердца, но не исследованном в этом контексте. С помощью биохимических тестов связывания они показали, что BACH2 прикрепляется к консервативным участкам в промоторе MIAT — регуляторной области, управляющей продукцией MIAT. В человеческих сердечных фибробластах и кардиомиоцитах повышение уровня BACH2 снижало MIAT, тогда как инактивирование BACH2 повышало MIAT. Важно, что сердца пациентов с терминальной сердечной недостаточностью и сердца мышей после инфаркта демонстрировали ту же картину: уровни BACH2 были низкими при высоком MIAT. Лечение карведилолом у мышей повышало экспрессию BACH2 в зависимости от наличия β1‑рецепторов и β‑аррестин1, связывая поверхностный сигнал от препарата с этим ядерным переключателем генов.
Защита сердечных клеток от фиброза и гибели
Далее команда изучила, что именно делает BACH2 в поддерживающих и мышечных клетках сердца. В культивируемых человеческих кардиальных фибробластах потеря BACH2 усиливала маркеры фиброза, стимулировала пролиферацию клеток и повышала миграцию — поведение, способствующее накоплению рубцовой ткани — тогда как избыток BACH2 ослаблял эти фибротические признаки. В человеческих и мышиных кардиомиоцитах снижение BACH2 приводило к увеличению числа погибающих клеток и повышенной активности ферментов, связанных с гибелью, а также к снижению уровней белков, обеспечивающих выживание. Сверхэкспрессия BACH2 давала противоположный эффект, сохраняя выживание клеток при стрессе. В совокупности эти эксперименты поддерживают модель, в которой карведилол активирует β1‑рецепторы и β‑аррестин1, которые затем взаимодействуют с BACH2 в ядре, чтобы понизить MIAT и тем самым ограничить фиброз и потерю клеток.
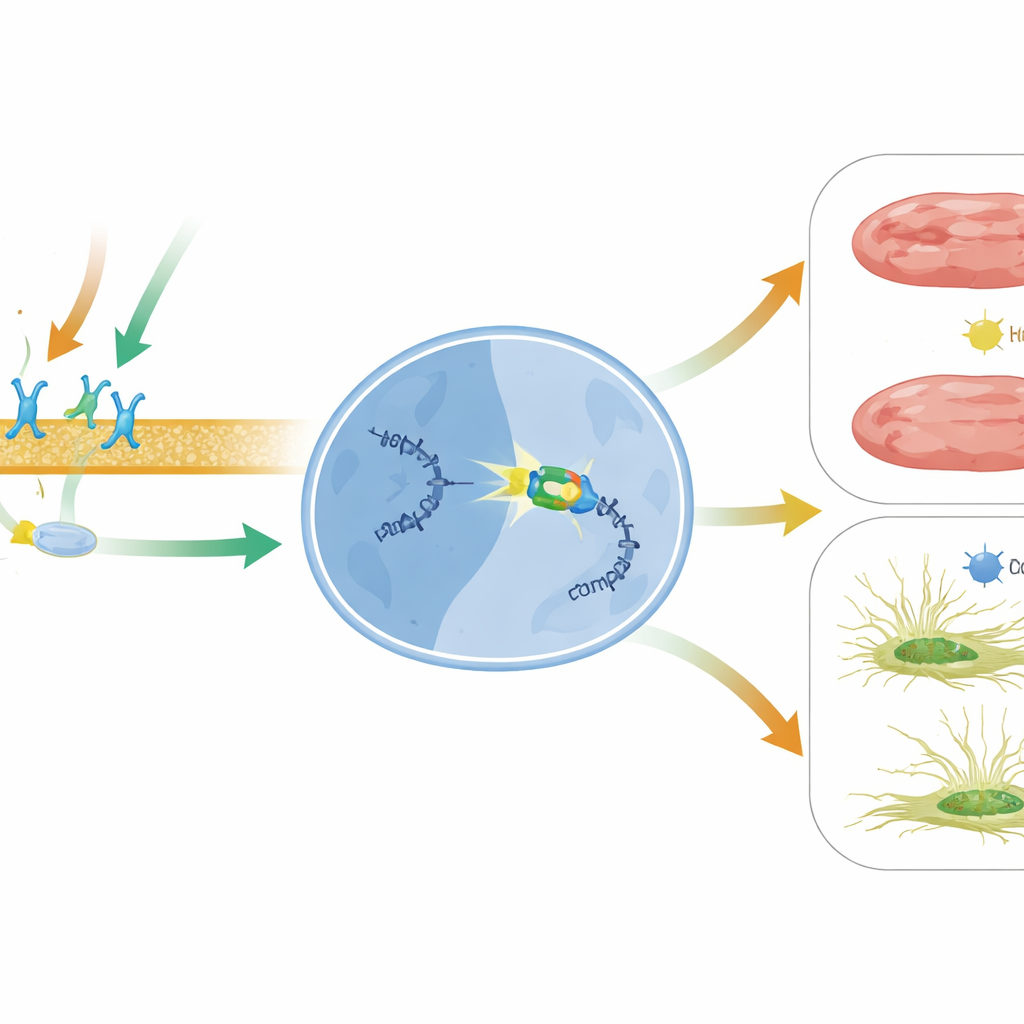
Что это может значить для будущего лечения сердца
Проще говоря, работа выявляет новую защитную ось внутри сердца с недостаточностью: активация β1‑рецептора и сигнальной каскады β‑аррестин1 под действием препарата включает BACH2, который затем отключает вредное «сообщение» MIAT, способствующее образованию рубцов и гибели клеток. Поскольку MIAT повышен при нескольких сердечных заболеваниях у людей и даже циркулирует в крови в качестве потенциального маркера повреждения сердца, нацеливание на путь BACH2–MIAT может дополнить существующие терапии. Стратегии, усиливающие активность BACH2, тонко настраивающие смещенную в сторону β‑аррестин1 сигнальную активность препаратами, подобными карведилолу, или непосредственно подавляющие MIAT с помощью РНК‑ориентированных инструментов, возможно, однажды помогут предотвратить ожесточение и прогрессирование сердечной недостаточности.
Цитирование: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Ключевые слова: сердечная недостаточность, кардиальный фиброз, длинная некодирующая РНК, терапия бета-блокаторами, апоптоз кардиомиоцитов