Clear Sky Science · pl
BACH2 łączy sygnalizację receptora β1-adrenergicznego/β-arrestyny1 z MIAT, hamując aktywację fibroblastów serca i apoptozę kardiomiocytów
Dlaczego to badanie serca ma znaczenie
Zawały i przewlekła niewydolność serca wciąż należą do głównych przyczyn zgonów, częściowo dlatego, że uszkodzona tkanka mięśnia sercowego jest zastępowana blizną, a żywotne komórki serca obumierają. To badanie odkrywa wcześniej nieznany wewnętrzny wyłącznik bezpieczeństwa w komórkach serca, który jest uruchamiany przez powszechnie stosowany lek beta‑adrenolityczny i w konsekwencji blokuje szkodliwy sygnał RNA związany z bliznowaceniem i śmiercią komórek. Zrozumienie tej wbudowanej ścieżki ochronnej może wskazać drogę do bardziej precyzyjnych terapii, które pozwolą uszkodzonym sercom dłużej i skuteczniej pracować.
Szkodliwy komunikat w zawodzących sercach
Gdy część serca cierpi z powodu niedokrwienia, wiele pracujących komórek mięśniowych (kardiomiocytów) obumiera i zostaje zastąpionych sztywną blizną wytwarzaną przez komórki podporowe zwane fibroblastami. Wcześniejsze badania zidentyfikowały długie niekodujące RNA zwane MIAT, które działa jak toksyczny komunikat: zwiększa ekspresję genów sprzyjających bliznowaceniu i zaprogramowanej śmierci komórkowej. Poziomy MIAT rosną w kilku chorobach serca u ludzi i w wielu modelach zwierzęcych uszkodzenia serca, a zablokowanie MIAT u myszy poprawia funkcję serca po zawale. Do tej pory jednak naukowcy nie wiedzieli, co reguluje samą MIAT ani jak standardowe leki sercowe mogą na nią wpływać.
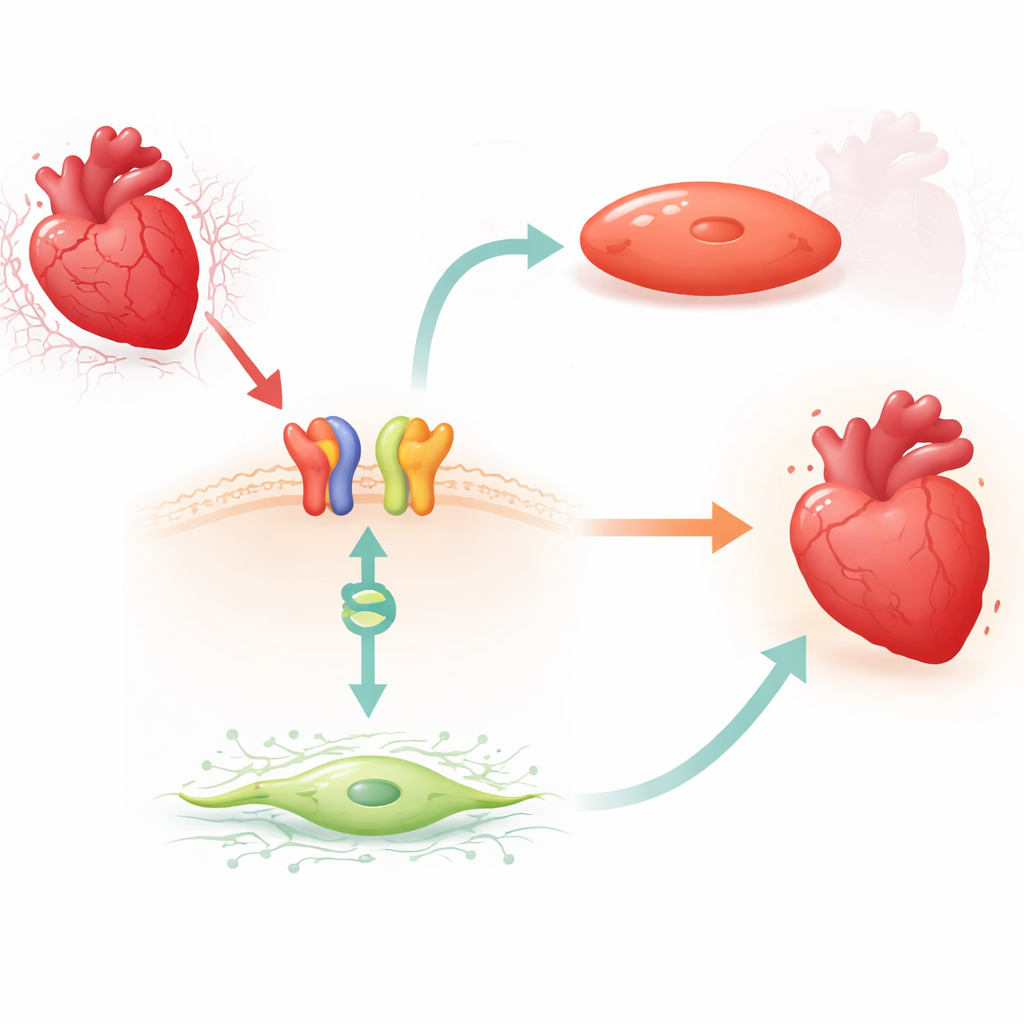
Powiązanie od sygnału powierzchniowego do przełącznika genowego aktywowane przez lek
Naukowcy skoncentrowali się na karwedilolu, szeroko stosowanym β-blokerze, który może uruchamiać ochronną ścieżkę sygnalizacyjną przez receptor β1-adrenergiczny i białko adaptujące zwane β-arrestyną1. U myszy stwierdzili, że krótkie leczenie karwedilolem konsekwentnie obniżało poziomy MIAT w lewej komorze, ale tylko wtedy, gdy obecne były receptory β1 i β-arrestyna1. Podobne spadki MIAT obserwowano w dorosłych ludzkich fibroblastach serca oraz w kardiomiocytach ludzkich i gryzoni wystawionych na laboratoryjny model niedotlenienia i ponownego utlenowania. We wszystkich tych systemach stres zwiększał MIAT, podczas gdy karwedilol odwracał ten wzrost, co sugeruje, że lek wykorzystuje naturalny hamulec na ten szkodliwy RNA.
Białko‑strażnik, które ucisza szkodliwe RNA
Głębiej zespół poszukiwał białka wiążącego DNA, które mogłoby współpracować z β-arrestyną1 w jądrze komórkowym. Skoncentrowali się na BACH2, czynniku transkrypcyjnym wcześniej powiązanym z ochroną przed innymi formami uszkodzenia serca, ale nie badanym w tym kontekście. Przy użyciu biochemicznych testów wiązania wykazali, że BACH2 przyłącza się do zachowanych odcinków w promotorze MIAT, czyli regionie kontrolnym napędzającym produkcję MIAT. W ludzkich fibroblastach serca i kardiomiocytach zwiększenie BACH2 zmniejszało poziomy MIAT, podczas gdy wyciszenie BACH2 podnosiło MIAT. Co istotne, serca pacjentów z zaawansowaną niewydolnością serca oraz serca myszy po zawale wykazywały ten sam wzorzec: poziomy BACH2 były niskie, gdy MIAT była wysoka. Leczenie karwedilolem u myszy podnosiło ekspresję BACH2 w sposób zależny od β1 i β-arrestyny1, łącząc sygnał z powierzchni komórki z tym jądrowym przełącznikiem genowym.
Ochrona komórek serca przed bliznowaceniem i śmiercią
Zespół następnie sprawdził, co BACH2 robi w komórkach podporowych i mięśniowych serca. W hodowlach ludzkich fibroblastów serca utrata BACH2 nasilała markery włóknienia, zwiększała proliferację komórek i poprawiała ich migrację — zachowania sprzyjające gromadzeniu się blizny — podczas gdy dodatkowy BACH2 tłumił te cechy fibrotczne. W kardiomiocytach ludzkich i mysich obniżenie BACH2 prowadziło do większej liczby umierających komórek i wyższej aktywności enzymów związanych ze śmiercią, wraz ze zmniejszeniem poziomów naturalnych białek wspierających przeżycie. Nadekspresja BACH2 działała odwrotnie, chroniąc komórki przed stresem. Razem te eksperymenty wspierają model, w którym karwedilol aktywuje receptory β1 i β-arrestynę1, które następnie współdziałają z BACH2 w jądrze, aby wyciszyć MIAT i w ten sposób ograniczyć bliznowacenie i utratę komórek.
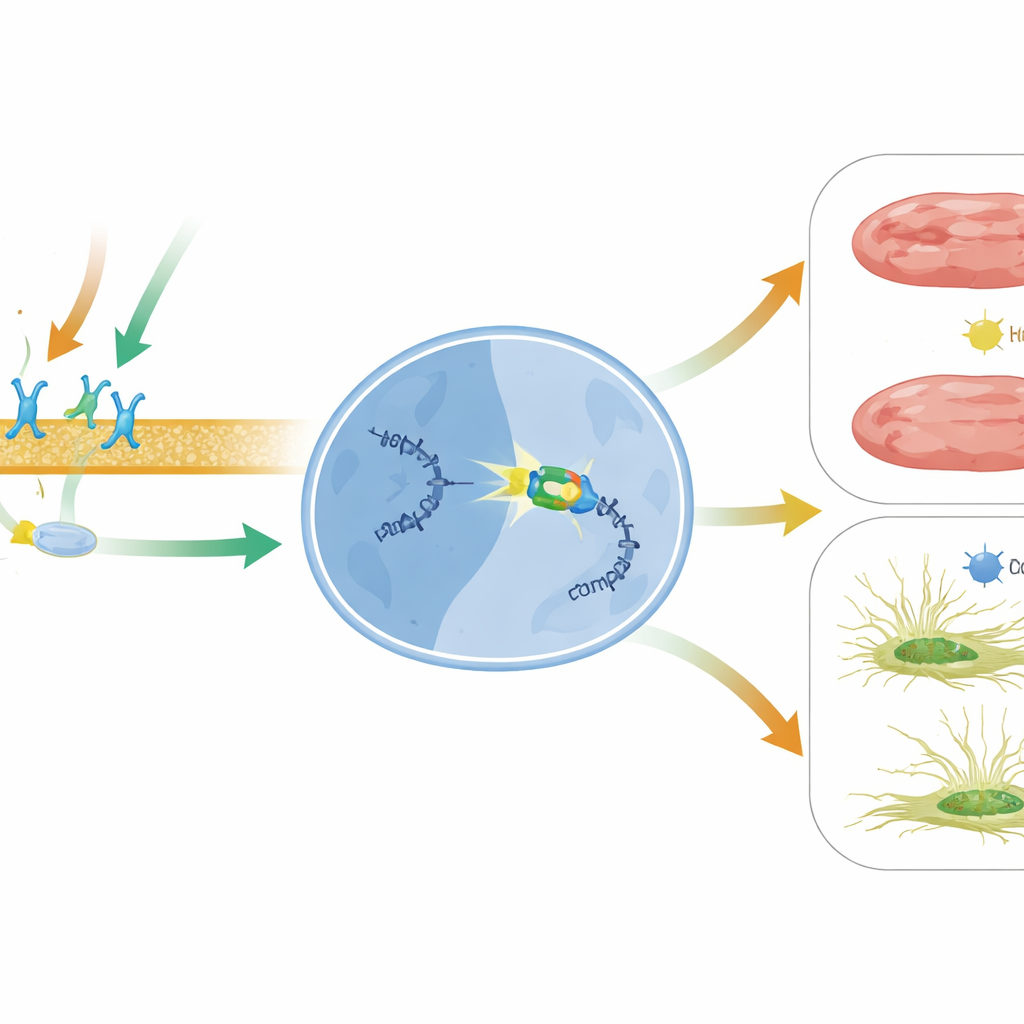
Co to może znaczyć dla przyszłych terapii sercowych
Mówiąc prosto, praca ta ujawnia nową osi ochronną wewnątrz zawodzącego serca: aktywacja receptora β1 i sygnalizacji zależnej od β-arrestyny1 przez lek włącza BACH2, który następnie wyłącza szkodliwy komunikat MIAT napędzający tworzenie blizny i śmierć komórek. Ponieważ MIAT jest podwyższone w kilku chorobach serca u ludzi i nawet krąży we krwi jako potencjalny marker uszkodzenia serca, celowanie w szlak BACH2–MIAT mogłoby uzupełniać istniejące terapie. Strategie zwiększające aktywność BACH2, dostrajające sygnalizację skierowaną na β-arrestynę1 za pomocą leków takich jak karwedilol, lub bezpośrednie wyciszanie MIAT narzędziami RNA mogłyby pewnego dnia pomóc zapobiegać usztywnianiu się i niewydolności uszkodzonych serc.
Cytowanie: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Słowa kluczowe: niewydolność serca, włóknienie serca, długa niekodująca RNA, terapia β-blokerami, apoptoza kardiomiocytów