Clear Sky Science · es
BACH2 conecta la señalización del receptor β1-adrenérgico/β-arrestina1 con MIAT para inhibir la activación de fibroblastos cardíacos y la apoptosis de cardiomiocitos
Por qué importa este estudio cardiaco
Los infartos y la insuficiencia cardíaca crónica siguen siendo causas principales de mortalidad, en parte porque el músculo cardíaco lesionado se reemplaza por tejido cicatricial y las células vitales del corazón mueren. Este estudio descubre un interruptor de seguridad hasta ahora desconocido dentro de las células cardíacas que se activa con un betabloqueante común y que, a su vez, apaga una señal de ARN dañina vinculada a la formación de cicatrices y la muerte celular. Comprender esta vía protectora innata podría señalar recursos para terapias más precisas que mantengan los corazones dañados funcionando por más tiempo y con mejor rendimiento.
Un mensaje dañino en los corazones en fallo
Cuando una parte del corazón se queda sin riego sanguíneo, muchas células musculares funcionales (cardiomiocitos) mueren y son reemplazadas por una cicatriz rígida fabricada por células de sostén llamadas fibroblastos. Investigaciones anteriores identificaron un ARN largo no codificante llamado MIAT que actúa como un mensaje tóxico: potencia genes que promueven la fibrosis y la muerte programada celular. Los niveles de MIAT aumentan en varias enfermedades cardíacas humanas y en múltiples modelos animales de lesión cardíaca, mientras que bloquear MIAT en ratones mejora la función del corazón tras un infarto. Sin embargo, hasta ahora no se sabía qué controla el propio MIAT ni cómo los fármacos estándar para el corazón podrían influir en él.
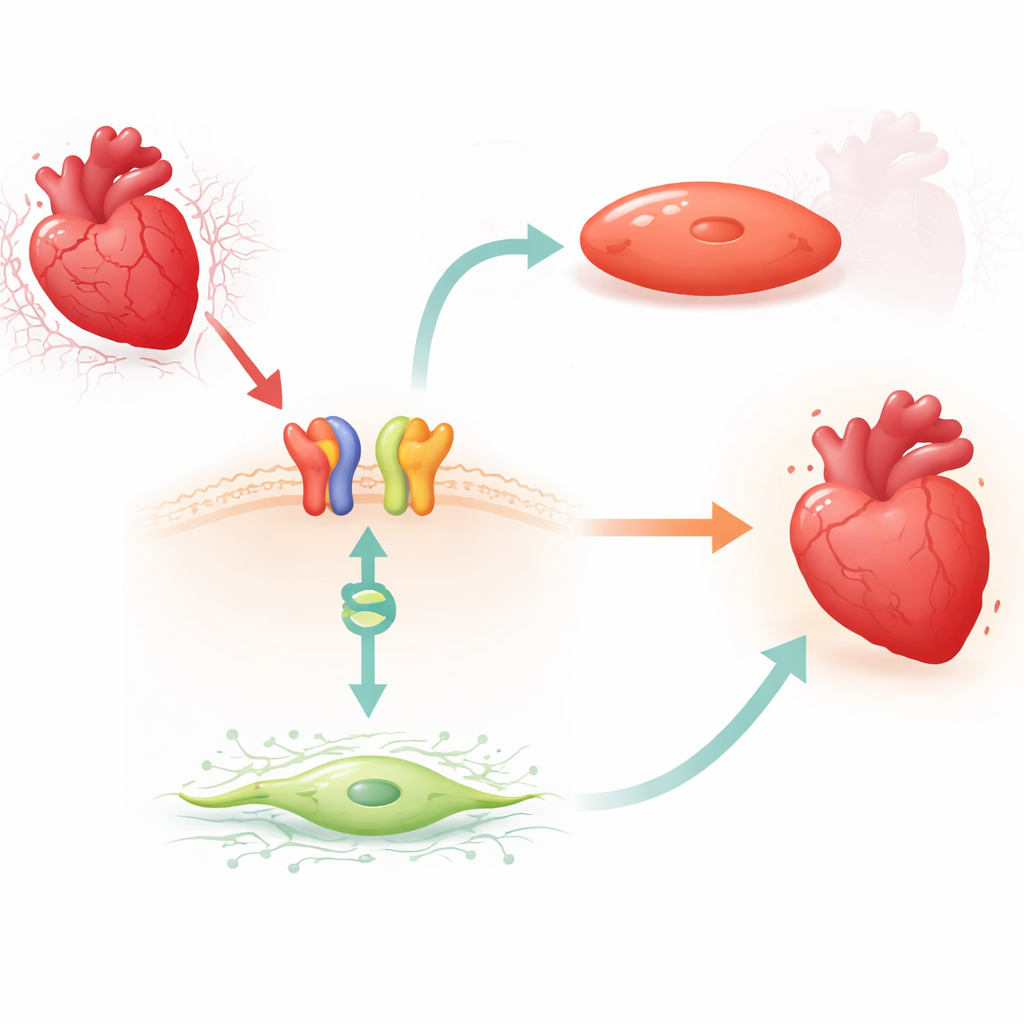
Un enlace activado por fármaco desde la señal de la superficie hasta el interruptor génico
Los investigadores se centraron en carvedilol, un betabloqueante de uso habitual que puede desencadenar una vía de señalización protectora a través del receptor β1-adrenérgico y una proteína adaptadora llamada β-arrestina1. En ratones, hallaron que un tratamiento breve con carvedilol redujo de forma consistente los niveles de MIAT en el ventrículo izquierdo, pero solo cuando estaban presentes los receptores β1 y la β-arrestina1. Reducciones similares de MIAT se observaron en fibroblastos cardíacos humanos adultos y en cardiomiocitos humanos y de roedores expuestos a un modelo de laboratorio de baja oxigenación y reoxigenación. En estos sistemas, el estrés aumentó MIAT, mientras que el carvedilol revirtió ese aumento, lo que sugiere que el fármaco aprovecha un freno natural sobre este ARN dañino.
La proteína guardiana que silencia el ARN dañino
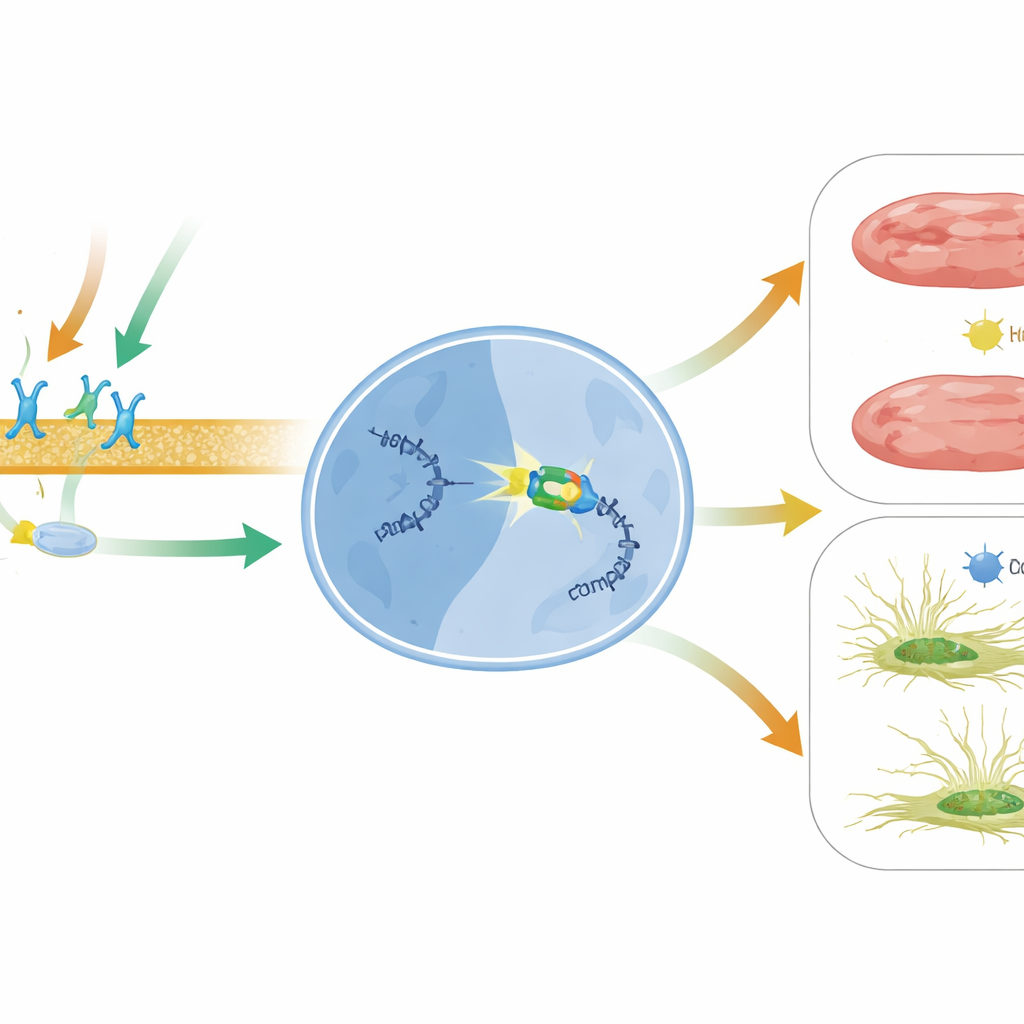
Profundizando, el equipo buscó una proteína que se una al ADN y que pudiera asociarse con β-arrestina1 en el núcleo celular. Se centraron en BACH2, un factor de transcripción vinculado previamente a la protección frente a otras formas de lesión cardíaca pero no estudiado en este contexto. Mediante ensayos bioquímicos de unión, mostraron que BACH2 se adhiere a tramos conservados en el promotor de MIAT, la región de control que impulsa la producción de MIAT. En fibroblastos cardíacos humanos y en cardiomiocitos, aumentar BACH2 redujo los niveles de MIAT, mientras que silenciar BACH2 elevó MIAT. De forma importante, corazones de pacientes con insuficiencia cardíaca avanzada y de ratones tras un infarto mostraron el mismo patrón: los niveles de BACH2 eran bajos cuando MIAT era alto. El tratamiento con carvedilol en ratones aumentó la expresión de BACH2 de manera dependiente de β1 y de β-arrestina1, vinculando la señal de superficie inducida por el fármaco con este interruptor génico nuclear.
Protección de las células cardíacas frente a la fibrosis y la muerte
El equipo preguntó luego qué hace BACH2 en las células de sostén y en las células musculares del corazón. En fibroblastos cardíacos humanos en cultivo, la pérdida de BACH2 incrementó marcadores de fibrosis, aumentó el crecimiento celular y potenció la migración celular—conductas que favorecen la acumulación de cicatriz—mientras que un exceso de BACH2 atenuó estos rasgos fibróticos. En cardiomiocitos humanos y de ratón, reducir BACH2 condujo a más células muertas y mayor actividad de enzimas relacionadas con la apoptosis, junto con niveles reducidos de proteínas de supervivencia natural. La sobreexpresión de BACH2 tuvo los efectos opuestos, preservando la supervivencia celular bajo estrés. En conjunto, estos experimentos respaldan un modelo en el que carvedilol activa los receptores β1 y la β-arrestina1, que luego cooperan con BACH2 en el núcleo para disminuir MIAT y, al hacerlo, limitar la fibrosis y la pérdida celular.
Qué podría significar esto para futuros tratamientos cardíacos
En términos sencillos, este trabajo revela un nuevo eje protector dentro del corazón en fallo: la señalización de receptor β1 y β-arrestina1 activada por fármacos enciende BACH2, que a su vez apaga el mensaje dañino MIAT que impulsa la formación de cicatrices y la muerte celular. Dado que MIAT está elevado en varias enfermedades cardíacas humanas e incluso circula en sangre como posible marcador de daño cardíaco, dirigir la vía BACH2–MIAT podría complementar las terapias actuales. Estrategias que aumenten la actividad de BACH2, ajusten la señalización sesgada hacia β-arrestina1 con fármacos como el carvedilol, o silencien directamente MIAT con herramientas basadas en ARN podrían algún día ayudar a evitar que los corazones dañados se endurezcan y fallen.
Cita: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Palabras clave: insuficiencia cardíaca, fibrosis cardíaca, ARN largo no codificante, terapia con betabloqueantes, apoptosis de cardiomiocitos