Clear Sky Science · nl
BACH2 koppelt β1-adrenerge receptor/β-arrestine1-signalerin g aan MIAT om activatie van cardiale fibroblasten en apoptose van cardiomyocyten te remmen
Waarom deze hartstudie ertoe doet
Hartaanvallen en chronisch hartfalen blijven belangrijke doodsoorzaken, deels omdat beschadigd hartweefsel wordt vervangen door littekenweefsel en vitale hartcellen afsterven. Deze studie ontdekt een eerder onbekende beveiligingsschakel binnen hartcellen die wordt geactiveerd door een veelgebruikt beta‑blokkerend geneesmiddel en die op zijn beurt een schadelijk RNA-signaal dat samenhangt met littekenvorming en celdood uitschakelt. Inzicht in deze ingebouwde beschermingsroute kan wijzen op preciezere therapieën die beschadigde harten langer en beter laten pompen.
Een schadelijke boodschap in falende harten
Wanneer een deel van het hart bloed tekortkomt, sterven veel werkende spiercellen (cardiomyocyten) en worden ze vervangen door stijf littekenweefsel dat door ondersteunende cellen, fibroblasten, wordt aangemaakt. Eerder onderzoek identificeerde een lang niet-coderend RNA genaamd MIAT dat als een toxisch bericht werkt: het verhoogt genen die littekenvorming en geprogrammeerde celdood bevorderen. MIAT-niveaus stijgen bij meerdere menselijke hartaandoeningen en in diverse diermodellen van hartbeschadiging, terwijl het blokkeren van MIAT in muizen de hartfunctie na een hartaanval verbetert. Tot nu toe wisten wetenschappers echter niet wat MIAT zelf reguleert of hoe gangbare hartmedicijnen het beïnvloeden.
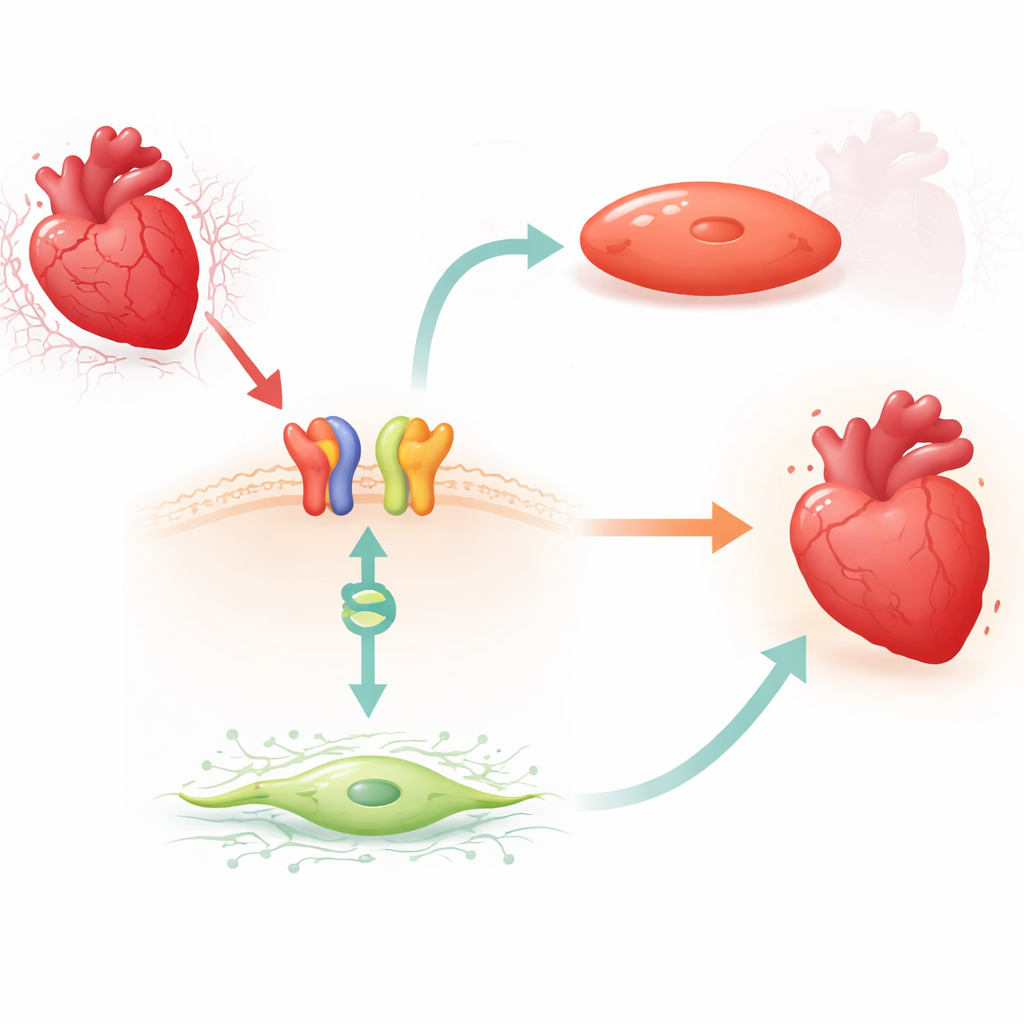
Een door geneesmiddel geactiveerde schakel van oppervlaksignaal naar genregulatie
De onderzoekers richtten zich op carvedilol, een veelgebruikte beta‑blokker die een beschermende signaalweg kan activeren via de beta1-adrenerge receptor en een adaptor-eiwit genaamd beta‑arrestine1. In muizen vonden ze dat korte behandeling met carvedilol consequent de MIAT-niveaus in het linkerventrikel verlaagde, maar alleen wanneer beta1-receptoren en beta‑arrestine1 aanwezig waren. Vergelijkbare MIAT‑verlagingen traden op in volwassen humane cardiale fibroblasten en in humane en knaagdier‑cardiomyocyten die werden blootgesteld aan een laboratoriummodel van zuurstoftekort en re-oxygenatie. In al deze systemen verhoogde stress MIAT, terwijl carvedilol deze stijging keerde, wat suggereert dat het geneesmiddel een natuurlijke rem op het schadelijke RNA aanspreekt.
Het waakzame eiwit dat het schadelijke RNA tot zwijgen brengt
Dieper gravend gingen de onderzoekers op zoek naar een DNA-bindend eiwit dat in de kern met beta‑arrestine1 zou kunnen samenwerken. Ze concentreerden zich op BACH2, een transcriptiefactor die eerder is gekoppeld aan bescherming tegen andere vormen van hartbeschadiging maar in deze context niet eerder is bestudeerd. Met biochemische bindingstests toonden ze aan dat BACH2 zich hecht aan geconserveerde sequenties in de MIAT-promoter, het controlegebied dat MIAT-productie aandrijft. In humane hartfibroblasten en cardiomyocyten verminderde versterking van BACH2 MIAT-niveaus, terwijl uitschakeling van BACH2 MIAT verhoogde. Belangrijk is dat harten van patiënten met gevorderd hartfalen en van muizen na een hartaanval hetzelfde patroon lieten zien: BACH2-niveaus waren laag wanneer MIAT hoog was. Carvedilolbehandeling bij muizen verhoogde BACH2-expressie op een beta1- en beta‑arrestine1‑afhankelijke manier, waarmee het oppervlaktesignaal van het geneesmiddel aan deze nucleus-genregelaar werd gekoppeld.
Bescherming van hartcellen tegen littekenvorming en celdood
Vervolgens vroegen de onderzoekers wat BACH2 precies doet in ondersteunende en spiercellen van het hart. In gekweekte humane cardiale fibroblasten leidde verlies van BACH2 tot hogere markers van fibrose, verhoogde celdeling en meer celmigratie — gedragingen die littekenopbouw bevorderen — terwijl extra BACH2 deze fibrotische kenmerken dempte. In menselijke en muis‑cardiomyocyten leidde verlaging van BACH2 tot meer stervende cellen en hogere activiteit van aan celdood gerelateerde enzymen, samen met verminderde niveaus van natuurlijke overlevingsproteïnen. Overexpressie van BACH2 had de tegengestelde effecten en bevorderde celsurvival onder stress. Samen ondersteunen deze experimenten een model waarin carvedilol beta1-receptoren en beta‑arrestine1 activeert, die vervolgens in de kern met BACH2 samenwerken om MIAT te remmen en zo littekenvorming en celverlies te beperken.
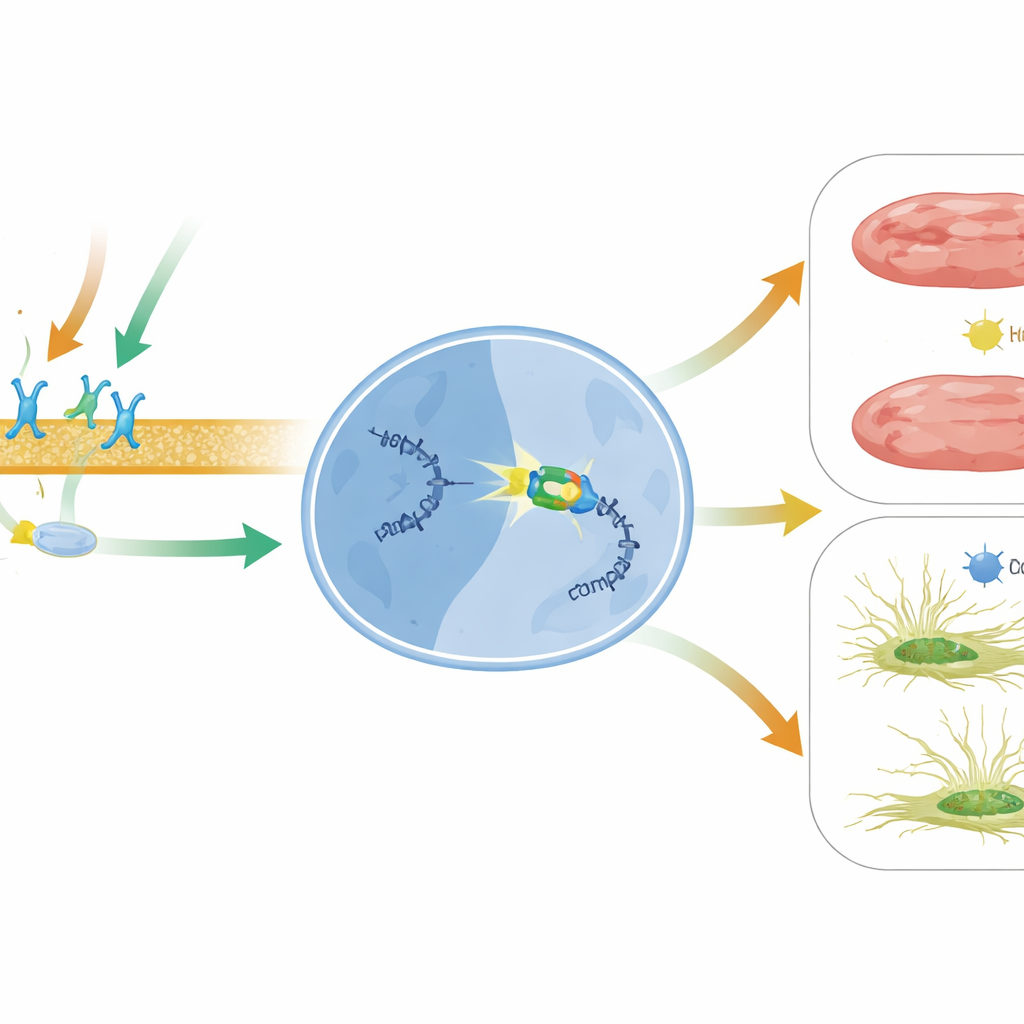
Wat dit kan betekenen voor toekomstige hartbehandelingen
In eenvoudige woorden onthult dit werk een nieuwe beschermende as in het falende hart: door geneesmiddel geactiveerde beta1-receptor- en beta‑arrestine1‑signaleringsroute schakelt BACH2 in, dat vervolgens het schadelijke MIAT‑bericht uitzet dat littekenvorming en celdood bevordert. Omdat MIAT verhoogd is bij verschillende menselijke hartaandoeningen en zelfs in het bloed circuleert als potentiële marker van hartschade, zou het richten op deze BACH2–MIAT-route een aanvulling kunnen vormen op huidige therapieën. Strategieën die BACH2-activiteit versterken, beta‑arrestine1‑georiënteerde signalering met geneesmiddelen zoals carvedilol verfijnen, of MIAT direct met RNA-gebaseerde middelen onderdrukken, kunnen op den duur helpen voorkomen dat beschadigde harten verstijven en falen.
Bronvermelding: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Trefwoorden: hartfalen, cardiale fibrose, lange niet-coderende RNA, beta-blokkertherapie, apoptose van cardiomyocyten