Clear Sky Science · it
BACH2 collega il segnale β1-adrenergico/β-arrestina1 a MIAT per inibire l’attivazione dei fibroblasti cardiaci e l’apoptosi dei cardiomiociti
Perché questo studio sul cuore è importante
Infarti e insufficienza cardiaca cronica restano tra le principali cause di morte, in parte perché il muscolo cardiaco lesionato viene sostituito da tessuto cicatriziale e le cellule vitali del cuore vanno perdute. Questo studio scopre un interruttore di sicurezza interno alle cellule cardiache che viene attivato da un comune beta‑bloccante e che, a sua volta, spegne un segnale RNA dannoso associato a fibrosi e morte cellulare. Capire questa via protettiva intrinseca potrebbe indicare la strada verso terapie più mirate che mantengano il cuore danneggiato a pompare più a lungo e meglio.
Un messaggio dannoso nei cuori in insufficienza
Quando una parte del cuore è privata di sangue, molti cardiomiociti muoiono e vengono rimpiazzati da una cicatrice rigida prodotta dalle cellule di supporto chiamate fibroblasti. Ricerche precedenti hanno identificato un lungo RNA non codificante chiamato MIAT che si comporta come un messaggio tossico: aumenta l’espressione di geni che promuovono la formazione di cicatrici e la morte cellulare programmata. I livelli di MIAT aumentano in diverse malattie cardiache umane e in molti modelli animali di danno cardiaco, mentre bloccare MIAT nei topi migliora la funzione cardiaca dopo un infarto. Tuttavia, fino ad ora non si sapeva cosa regolasse MIAT né come i farmaci cardiaci standard potessero influenzarlo.
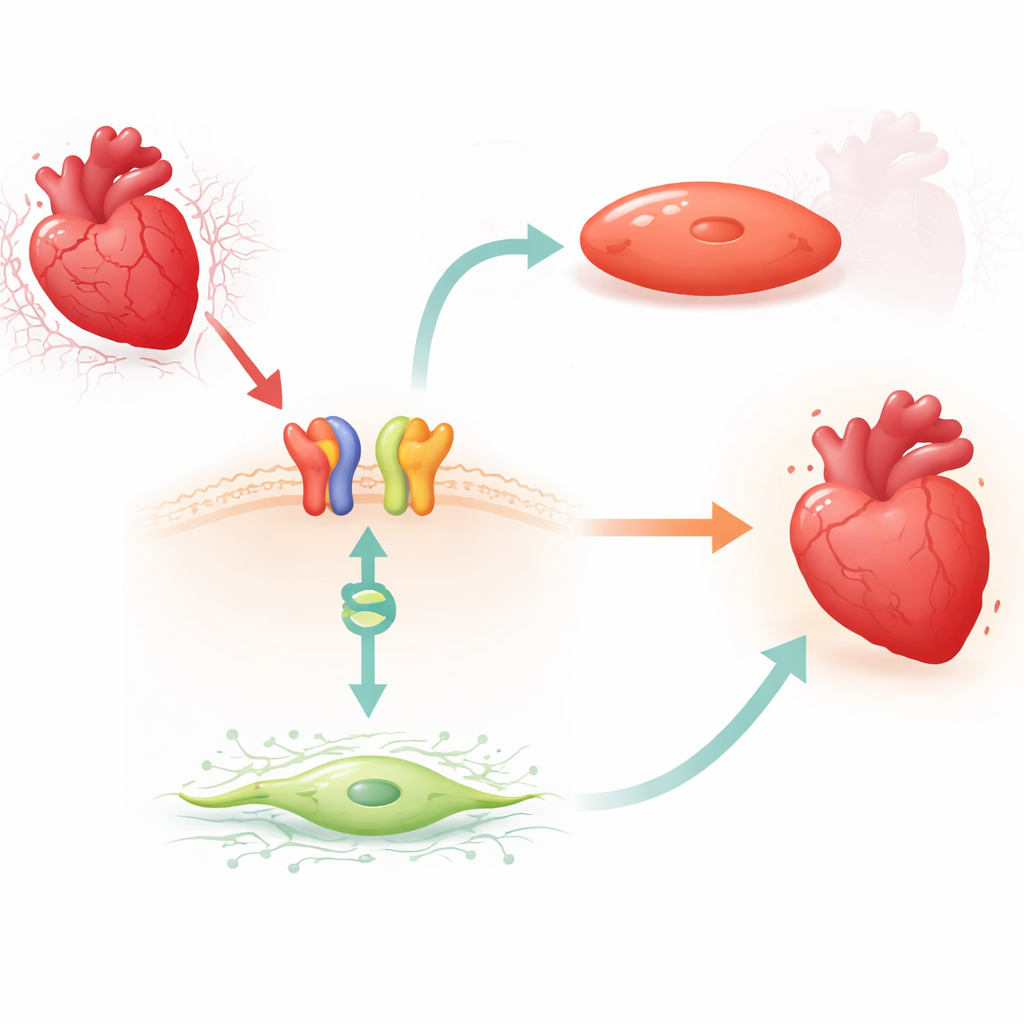
Un collegamento attivato da farmaco dal segnale di superficie all’interruttore genico
I ricercatori si sono concentrati sul carvedilolo, un beta‑bloccante ampiamente usato che può attivare una via protettiva attraverso il recettore β1‑adrenergico e una proteina adattatrice chiamata β‑arrestina1. Nei topi hanno osservato che un breve trattamento con carvedilolo riduceva costantemente i livelli di MIAT nel ventricolo sinistro, ma solo quando erano presenti i recettori β1 e la β‑arrestina1. Riduzioni simili di MIAT si sono verificate nei fibroblasti cardiaci umani adulti e nelle cellule miocardiche umane e di roditore esposte a un modello di laboratorio di ipossia e riossigenazione. In tutti questi sistemi, lo stress aumentava MIAT, mentre il carvedilolo invertiva questo aumento, suggerendo che il farmaco sfrutti un freno naturale sul RNA dannoso.
La proteina guardiana che zittisce l’RNA dannoso
Approfondendo, il gruppo ha cercato una proteina legante il DNA che potesse cooperare con la β‑arrestina1 nel nucleo cellulare. Si sono concentrati su BACH2, un fattore di trascrizione precedentemente collegato alla protezione contro altre forme di danno cardiaco ma non studiato in questo contesto. Tramite test biochimici di legame hanno mostrato che BACH2 si attacca a tratti conservati nel promotore di MIAT, la regione di controllo che regola la produzione di MIAT. In fibroblasti cardiaci umani e in cardiomiociti, aumentare BACH2 riduceva i livelli di MIAT, mentre silenziare BACH2 li aumentava. Importante, i cuori di pazienti con insufficienza cardiaca avanzata e quelli di topi dopo infarto mostravano lo stesso schema: i livelli di BACH2 erano bassi quando MIAT era elevato. Il trattamento con carvedilolo nei topi aumentava l’espressione di BACH2 in modo dipendente da β1 e β‑arrestina1, collegando il segnale di superficie del farmaco a questo interruttore genico nucleare.
Proteggere le cellule cardiache da fibrosi e morte
Il gruppo ha quindi chiesto cosa fa concretamente BACH2 alle cellule di supporto e alle cellule muscolari cardiache. In fibroblasti cardiaci umani coltivati, la perdita di BACH2 aumentava i marcatori di fibrosi, promuoveva la proliferazione cellulare e incrementava la migrazione cellulare — comportamenti che favoriscono l’accumulo di cicatrice — mentre un eccesso di BACH2 attenuava questi tratti fibrotici. In cardiomiociti umani e murini, la riduzione di BACH2 portava a più cellule morenti e a una maggiore attività di enzimi legati alla morte cellulare, insieme a livelli ridotti di proteine di sopravvivenza naturali. L’iperespressione di BACH2 produceva effetti opposti, preservando la sopravvivenza cellulare sotto stress. Complessivamente, questi esperimenti supportano un modello in cui il carvedilolo attiva i recettori β1 e la β‑arrestina1, che poi cooperano con BACH2 nel nucleo per abbassare MIAT e, così facendo, limitare la fibrosi e la perdita cellulare.
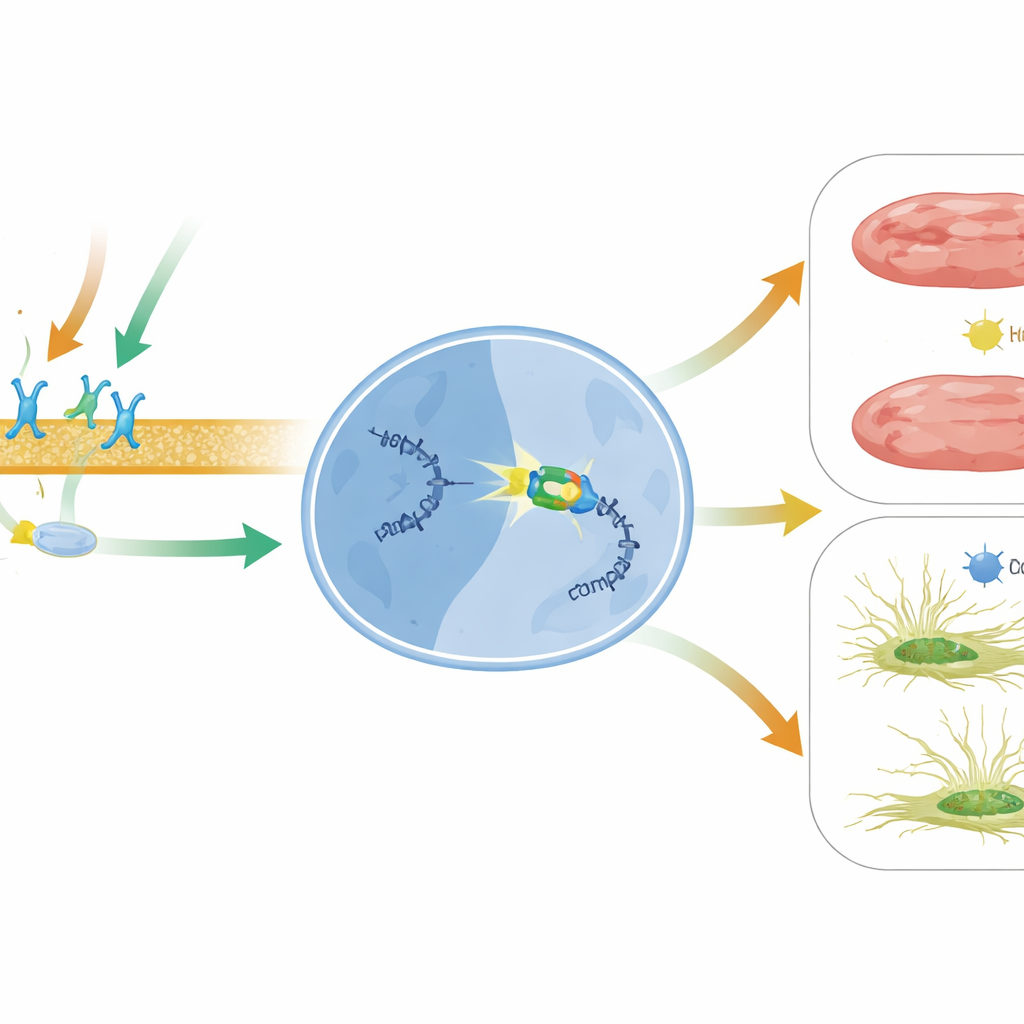
Cosa potrebbe significare per i trattamenti cardiaci futuri
In termini semplici, questo lavoro rivela un nuovo asse protettivo all’interno del cuore in insufficienza: il segnale indotto dal farmaco attraverso il recettore β1 e la β‑arrestina1 attiva BACH2, che poi spegne il messaggio dannoso MIAT che promuove la formazione di cicatrici e la morte cellulare. Poiché MIAT è elevato in diverse malattie cardiache umane e può persino circolare nel sangue come potenziale marcatore di danno cardiaco, mirare a questa via BACH2–MIAT potrebbe integrare le terapie attuali. Strategie che potenzino l’attività di BACH2, modulino in modo mirato il segnale bias‑β‑arrestina1 con farmaci come il carvedilolo, o silenzino direttamente MIAT con strumenti basati su RNA potrebbero un giorno contribuire a mantenere i cuori danneggiati meno rigidi e meno soggetti a insufficienza.
Citazione: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Parole chiave: insufficienza cardiaca, fibrosi cardiaca, RNA lungo non codificante, terapia con beta-bloccanti, apoptosi dei cardiomiociti