Clear Sky Science · fr
BACH2 relie le signal β1-adrénergique/β-arrestine1 à MIAT pour inhiber l’activation des fibroblastes cardiaques et l’apoptose des cardiomyocytes
Pourquoi cette étude cardiaque est importante
Les infarctus et l’insuffisance cardiaque chronique restent des causes majeures de décès, en partie parce que le muscle cardiaque lésé est remplacé par du tissu cicatriciel et que des cellules cardiaques vitales meurent. Cette étude révèle un interrupteur de sécurité intracellulaire jusqu’alors inconnu, activé par un bêta‑bloquant courant qui, à son tour, éteint un signal ARN délétère lié à la cicatrisation et à la mort cellulaire. Comprendre cette voie protectrice intrinsèque pourrait orienter vers des thérapies plus précises permettant aux cœurs abîmés de fonctionner plus longtemps et mieux.
Un message nocif dans les cœurs en défaillance
Quand une région du cœur manque d’oxygène, de nombreux cardiomyocytes meurent et sont remplacés par une cicatrice rigide produite par des cellules de soutien appelées fibroblastes. Des travaux antérieurs ont identifié un ARN long non codant nommé MIAT qui agit comme un message toxique : il stimule des gènes favorisant la fibrose et l’apoptose. Les niveaux de MIAT augmentent dans plusieurs maladies cardiaques humaines et dans divers modèles animaux de lésion cardiaque, tandis que bloquer MIAT chez la souris améliore la fonction cardiaque après un infarctus. Jusqu’à présent, on ignorait toutefois ce qui régule MIAT lui‑même et comment les médicaments cardiaques standards peuvent l’influencer.
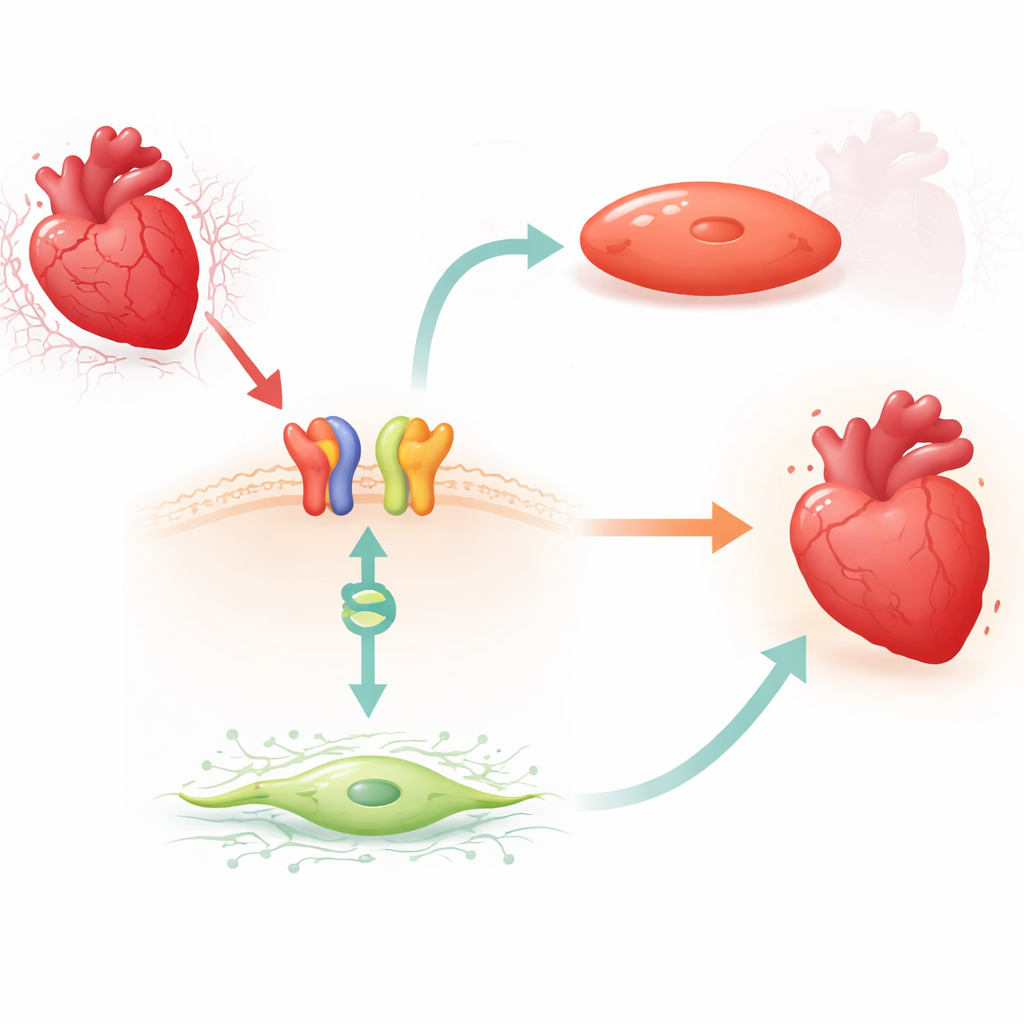
Un lien activé par un médicament entre signal de surface et interrupteur génétique
Les chercheurs se sont concentrés sur le carvedilol, un bêta‑bloquant largement utilisé capable d’engager une voie signalétique protectrice via le récepteur β1‑adrénergique et une protéine adaptatrice appelée β‑arrestine1. Chez la souris, ils ont observé qu’un traitement bref au carvedilol diminuait systématiquement les niveaux de MIAT dans le ventricule gauche, mais seulement en présence des récepteurs β1 et de la β‑arrestine1. Des réductions similaires de MIAT ont été constatées dans des fibroblastes cardiaques humains adultes et dans des cardiomyocytes humains et de rongeur exposés à un modèle expérimental d’hypoxie‑reoxygénation. Dans tous ces systèmes, le stress augmentait MIAT, tandis que le carvedilol inversait cette hausse, suggérant que le médicament exploite un frein naturel sur cet ARN nuisible.
La protéine « gardienne » qui calme l’ARN nocif
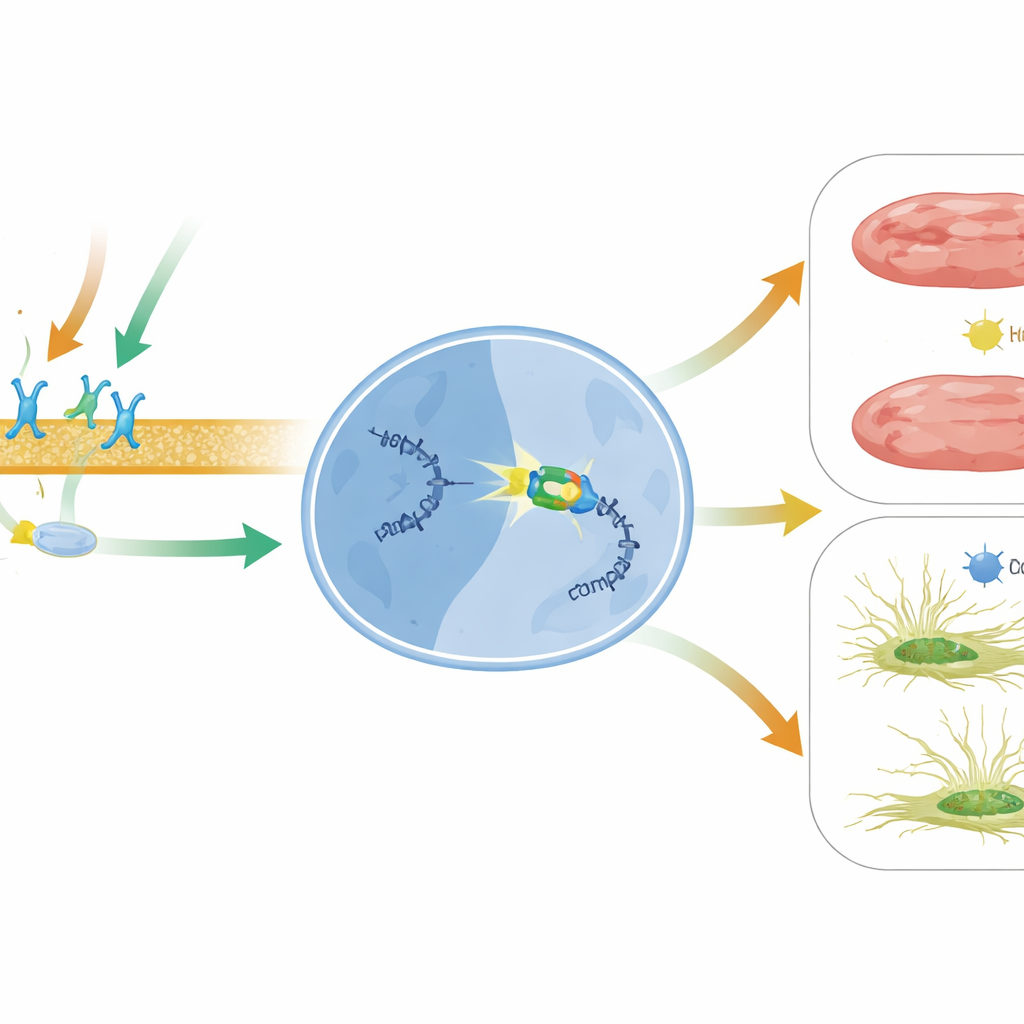
Pour approfondir, l’équipe a cherché une protéine liant l’ADN pouvant faire équipe avec la β‑arrestine1 dans le noyau. Ils ont ciblé BACH2, un facteur de transcription déjà associé à une protection contre d’autres formes de lésion cardiaque mais non étudié dans ce contexte. Par des tests biochimiques d’interaction, ils ont montré que BACH2 se fixe à des régions conservées du promoteur de MIAT, la région de contrôle qui pilote la production de MIAT. Dans les fibroblastes cardiaques humains et les cardiomyocytes, augmenter BACH2 réduisait MIAT, tandis que l’inactivation de BACH2 l’augmentait. De manière importante, les cœurs de patients en insuffisance cardiaque avancée et ceux de souris après infarctus présentaient le même profil : BACH2 était bas quand MIAT était élevé. Le traitement par carvedilol chez la souris augmentait l’expression de BACH2 de façon dépendante du β1 et de la β‑arrestine1, reliant ainsi le signal de surface au commutateur génique nucléaire.
Protéger les cellules cardiaques de la fibrose et de la mort
L’équipe a ensuite cherché à déterminer l’effet de BACH2 sur les cellules de soutien et musculaires cardiaques. Dans des fibroblastes cardiaques humains en culture, la perte de BACH2 augmentait les marqueurs de fibrose, la prolifération cellulaire et la migration—comportements favorisant l’accumulation de cicatrice—alors qu’un excès de BACH2 atténuait ces caractéristiques fibrosantes. Dans les cardiomyocytes humains et murins, la diminution de BACH2 entraînait davantage de cellules en voie de mort et une activité accrue d’enzymes liées à l’apoptose, avec des niveaux réduits de protéines de survie naturelles. La surexpression de BACH2 produisait l’effet inverse, préservant la survie cellulaire sous stress. Ensemble, ces expériences soutiennent un modèle dans lequel le carvedilol active les récepteurs β1 et la β‑arrestine1, qui coopèrent ensuite avec BACH2 dans le noyau pour réduire MIAT et, ce faisant, limiter la fibrose et la perte cellulaire.
Ce que cela pourrait signifier pour les traitements cardiaques futurs
En termes simples, ce travail révèle un nouvel axe protecteur dans le cœur en défaillance : le signal déclenché par le médicament via le récepteur β1 et la β‑arrestine1 active BACH2, qui éteint ensuite le message nocif MIAT qui favorise la formation de cicatrice et la mort cellulaire. Étant donné que MIAT est élevé dans plusieurs maladies cardiaques humaines et circule même dans le sang comme marqueur potentiel de lésion cardiaque, cibler la voie BACH2–MIAT pourrait compléter les thérapies actuelles. Des stratégies visant à renforcer l’activité de BACH2, à moduler finement le signal biaisé de la β‑arrestine1 avec des médicaments comme le carvedilol, ou à silencier directement MIAT par des outils à base d’ARN pourraient, un jour, aider à empêcher les cœurs endommagés de se rigidifier et de s’épuiser.
Citation: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Mots-clés: insuffisance cardiaque, fibrose cardiaque, ARN long non codant, traitement bêta-bloquant, apoptose des cardiomyocytes