Clear Sky Science · en
BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis
Why this heart study matters
Heart attacks and chronic heart failure remain leading causes of death, in part because injured heart muscle is replaced by scar tissue and vital heart cells die off. This study uncovers a previously unknown safety switch inside heart cells that is turned on by a common beta‑blocker drug and, in turn, shuts down a harmful RNA signal linked to scarring and cell death. Understanding this built‑in protective pathway could point the way to more precise therapies that keep damaged hearts pumping longer and better.
A harmful message in failing hearts
When part of the heart is starved of blood, many working muscle cells (cardiomyocytes) die and are replaced by stiff scar made by support cells called fibroblasts. Earlier research identified a long noncoding RNA called MIAT that acts like a toxic message: it boosts genes that promote scarring and programmed cell death. MIAT levels rise in several human heart diseases and in multiple animal models of heart injury, while blocking MIAT in mice improves heart function after a heart attack. Yet, until now, scientists did not know what controls MIAT itself or how standard heart drugs might influence it.
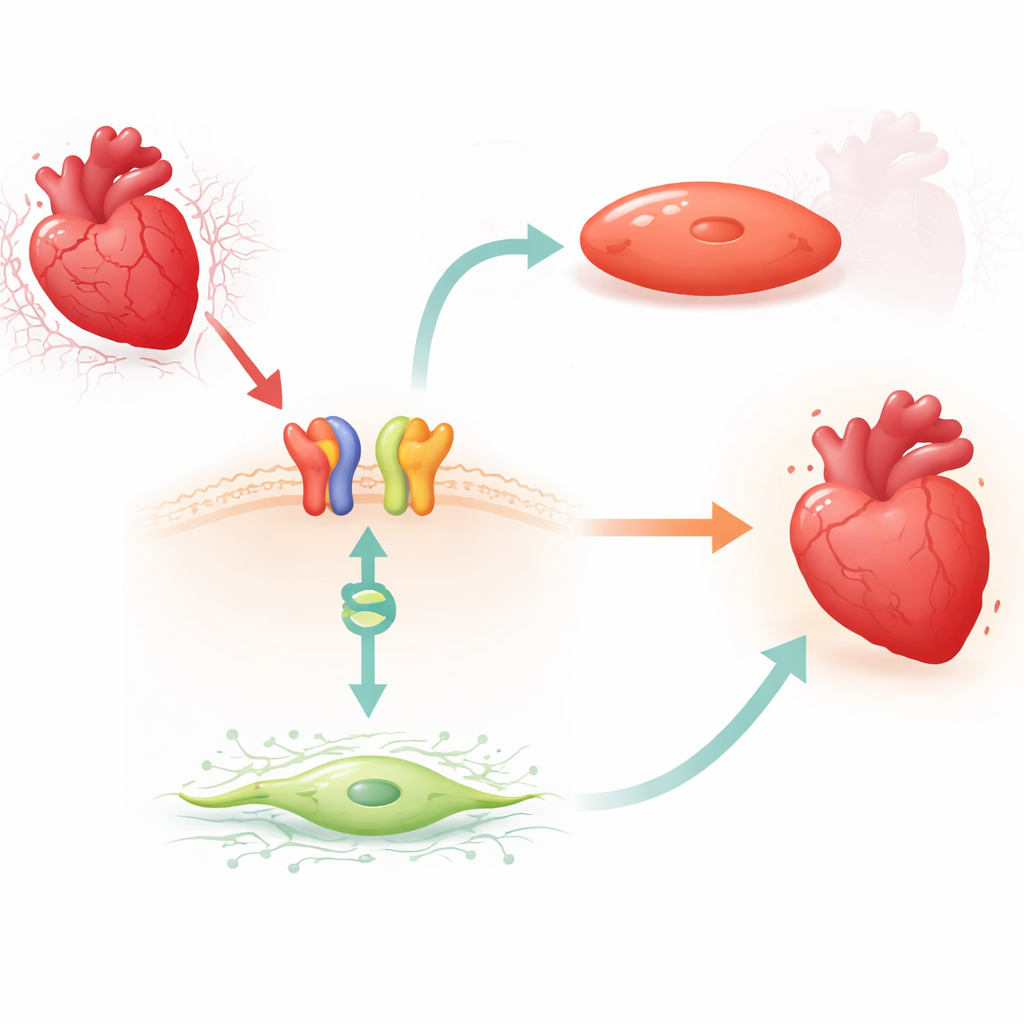
A drug‑activated link from surface signal to gene switch
The researchers focused on carvedilol, a widely used beta‑blocker that can trigger a protective signaling route through the beta1‑adrenergic receptor and an adaptor protein called beta‑arrestin1. In mice, they found that short carvedilol treatment consistently lowered MIAT levels in the left ventricle, but only when beta1 receptors and beta‑arrestin1 were present. Similar MIAT reductions occurred in adult human cardiac fibroblasts and in human and rodent heart muscle cells exposed to a laboratory model of low oxygen and re‑oxygenation. Across these systems, stress increased MIAT, while carvedilol reversed this rise, suggesting that the drug taps into a natural brake on the harmful RNA.
The guardian protein that quiets the harmful RNA
Diving deeper, the team looked for a DNA‑binding protein that could partner with beta‑arrestin1 in the cell nucleus. They homed in on BACH2, a transcription factor previously linked to protection against other forms of heart injury but not studied in this context. Using biochemical binding tests, they showed that BACH2 attaches to conserved stretches in the MIAT promoter, the control region that drives MIAT production. In human heart fibroblasts and cardiomyocytes, boosting BACH2 reduced MIAT levels, while silencing BACH2 raised MIAT. Importantly, hearts from patients with advanced heart failure and from mice after heart attack showed the same pattern: BACH2 levels were low when MIAT was high. Carvedilol treatment in mice raised BACH2 expression in a beta1‑ and beta‑arrestin1‑dependent way, tying the surface drug signal to this nuclear gene switch.
Protecting heart cells from scarring and death
The team then asked what BACH2 actually does to heart support and muscle cells. In cultured human cardiac fibroblasts, loss of BACH2 boosted markers of fibrosis, increased cell growth, and enhanced cell migration—behaviors that favor scar buildup—while extra BACH2 dampened these fibrotic traits. In human and mouse cardiomyocytes, lowering BACH2 led to more dying cells and higher activity of death‑related enzymes, along with reduced levels of natural survival proteins. Overexpressing BACH2 had the opposite effects, preserving cell survival under stress. Together, these experiments support a model in which carvedilol activates beta1 receptors and beta‑arrestin1, which then cooperate with BACH2 in the nucleus to turn down MIAT and, in doing so, limit scarring and cell loss.
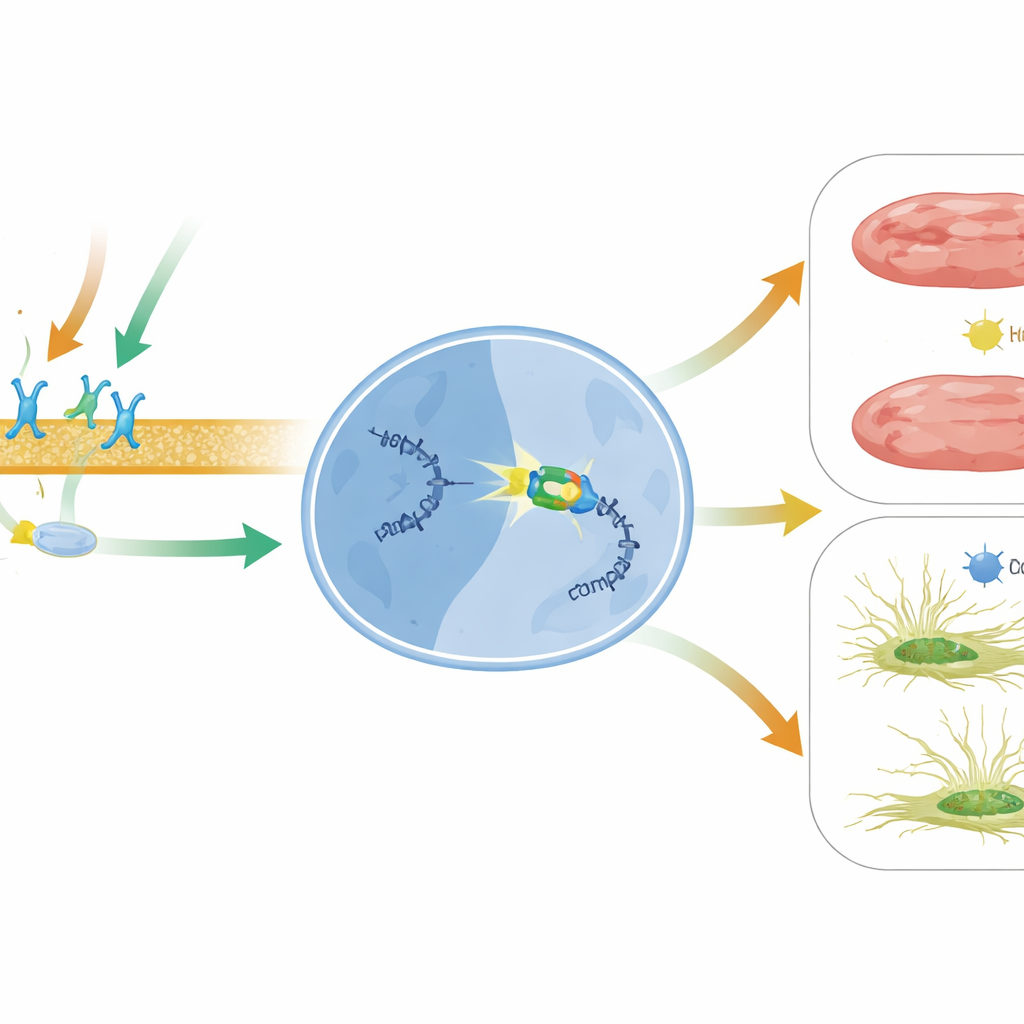
What this could mean for future heart treatments
In simple terms, this work reveals a new protective axis inside the failing heart: drug‑triggered beta1 receptor and beta‑arrestin1 signaling switches on BACH2, which then turns off the harmful MIAT message that pushes scar formation and cell death. Because MIAT is elevated in several human heart diseases and even circulates in the blood as a potential marker of heart damage, targeting this BACH2–MIAT pathway could complement current therapies. Strategies that enhance BACH2 activity, fine‑tune beta‑arrestin1‑biased signaling with drugs like carvedilol, or directly silence MIAT with RNA‑based tools may one day help keep damaged hearts from stiffening and failing.
Citation: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Keywords: heart failure, cardiac fibrosis, long noncoding RNA, beta-blocker therapy, cardiomyocyte apoptosis