Clear Sky Science · pt
BACH2 conecta o sinal β1-adrenérgico/β-arrestina1 à MIAT para inibir a ativação de fibroblastos cardíacos e a apoptose de cardiomiócitos
Por que este estudo cardíaco é importante
Infartos e insuficiência cardíaca crônica continuam sendo causas principais de morte, em parte porque o músculo cardíaco lesionado é substituído por tecido cicatricial e células cardíacas vitais morrem. Este estudo revela um interruptor de segurança até então desconhecido dentro das células cardíacas que é ativado por um beta‑bloqueador comum e, por sua vez, desliga um sinal de RNA prejudicial ligado à formação de cicatriz e à morte celular. Entender essa via de proteção intrínseca pode apontar para terapias mais precisas que mantenham corações danificados bombeando por mais tempo e com melhor função.
Uma mensagem nociva em corações em falência
Quando parte do coração fica sem suprimento sanguíneo, muitas células musculares funcionais (cardiomiócitos) morrem e são substituídas por uma cicatriz rígida produzida por células de suporte chamadas fibroblastos. Pesquisas anteriores identificaram um RNA longo não codificante chamado MIAT que age como uma mensagem tóxica: ele aumenta genes que promovem fibrose e morte celular programada. Os níveis de MIAT aumentam em várias doenças cardíacas humanas e em múltiplos modelos animais de lesão cardíaca, enquanto bloquear MIAT em camundongos melhora a função cardíaca após um infarto. Ainda assim, até agora, não se sabia o que controla o próprio MIAT ou como medicamentos padrão para o coração podem influenciá‑lo.
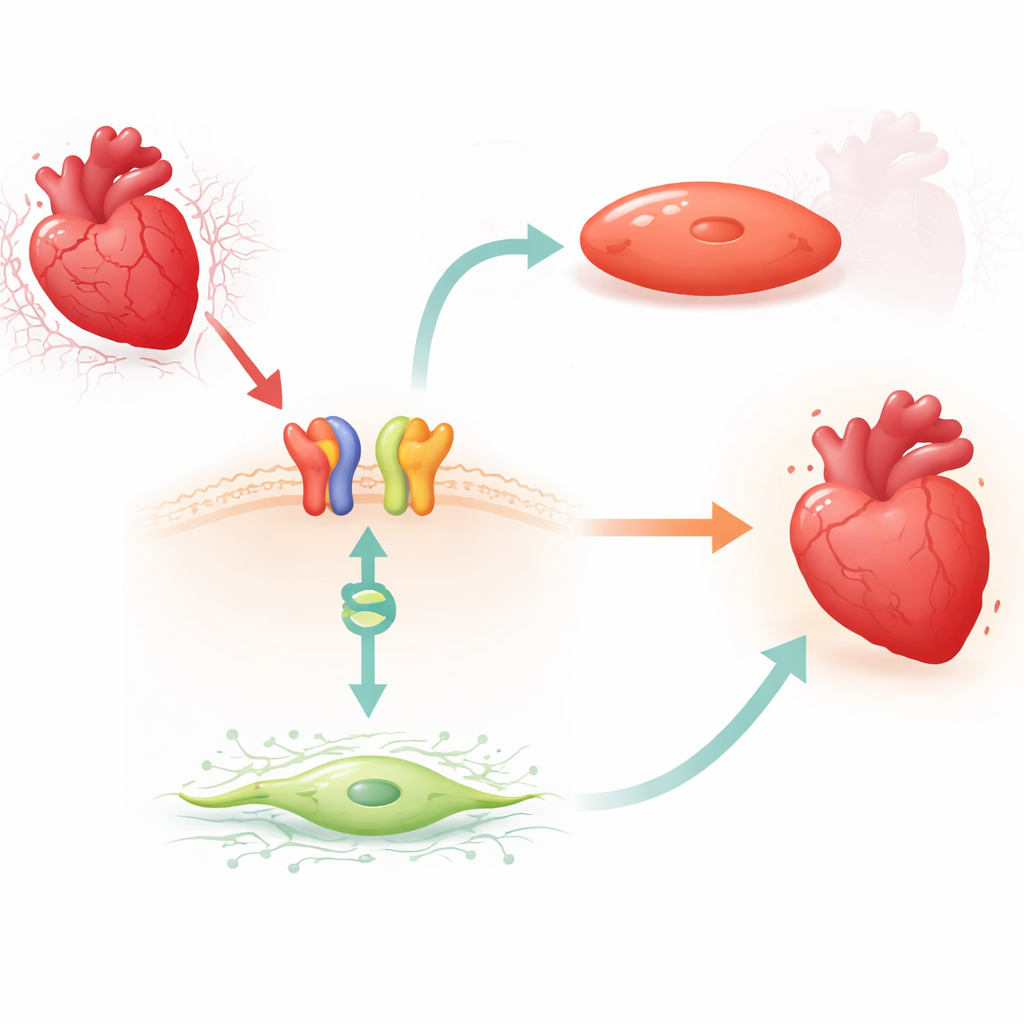
Uma ligação ativada por droga do sinal de superfície ao interruptor gênico
Os pesquisadores concentraram‑se no carvedilol, um beta‑bloqueador amplamente usado que pode ativar uma via de sinalização protetora através do receptor β1-adrenérgico e de uma proteína adaptadora chamada β‑arrestina1. Em camundongos, eles descobriram que um tratamento curto com carvedilol reduziu consistentemente os níveis de MIAT no ventrículo esquerdo, mas somente quando os receptores β1 e a β‑arrestina1 estavam presentes. Reduções semelhantes de MIAT ocorreram em fibroblastos cardíacos humanos adultos e em cardiomiócitos de humanos e roedores expostos a um modelo laboratorial de hipóxia seguida de reoxigenação. Nesses sistemas, o estresse aumentou o MIAT, enquanto o carvedilol reverteu esse aumento, sugerindo que o medicamento aproveita um freio natural sobre esse RNA nocivo.
A proteína guardiã que silencia o RNA prejudicial
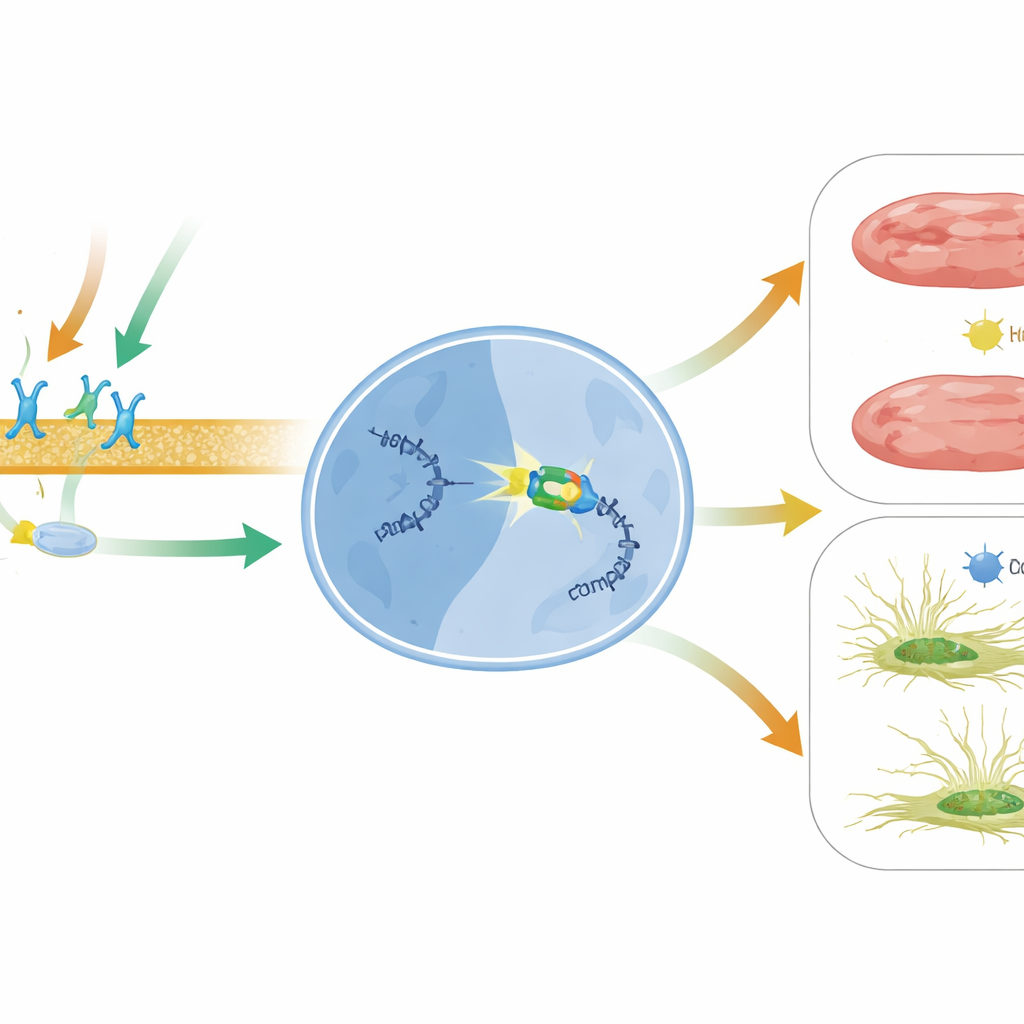
Aprofundando, a equipe procurou uma proteína ligada ao DNA que pudesse atuar em parceria com a β‑arrestina1 no núcleo celular. Eles se concentraram em BACH2, um fator de transcrição previamente associado à proteção contra outras formas de lesão cardíaca, mas não estudado nesse contexto. Usando testes bioquímicos de ligação, demonstraram que BACH2 se liga a trechos conservados no promotor de MIAT, a região de controle que impulsiona a produção de MIAT. Em fibroblastos e cardiomiócitos humanos, aumentar BACH2 reduziu os níveis de MIAT, enquanto silenciar BACH2 elevou o MIAT. Importante, corações de pacientes com insuficiência cardíaca avançada e de camundongos após infarto mostraram o mesmo padrão: níveis baixos de BACH2 quando MIAT estava alto. O tratamento com carvedilol em camundongos aumentou a expressão de BACH2 de forma dependente de β1 e de β‑arrestina1, ligando o sinal de superfície do medicamento a esse interruptor gênico nuclear.
Protegendo as células cardíacas da fibrose e da morte
A equipe então investigou o que BACH2 faz às células de suporte e musculares do coração. Em fibroblastos cardíacos humanos cultivados, a perda de BACH2 aumentou marcadores de fibrose, estimulou a proliferação celular e intensificou a migração celular — comportamentos que favorecem o acúmulo de cicatriz — enquanto excesso de BACH2 atenuou essas características fibróticas. Em cardiomiócitos humanos e de camundongo, reduzir BACH2 levou a mais células morrendo e maior atividade de enzimas relacionadas à morte, além de níveis reduzidos de proteínas naturais de sobrevivência. A superexpressão de BACH2 teve efeitos opostos, preservando a sobrevivência celular sob estresse. Juntos, esses experimentos apoiam um modelo no qual o carvedilol ativa receptores β1 e β‑arrestina1, que então cooperam com BACH2 no núcleo para reduzir MIAT e, ao fazer isso, limitar a fibrose e a perda celular.
O que isso pode significar para tratamentos cardíacos futuros
Em termos simples, este trabalho revela um novo eixo protetor dentro do coração em falência: a sinalização do receptor β1 e da β‑arrestina1 ativada por droga liga BACH2, que então desliga a mensagem nociva MIAT que impulsiona a formação de cicatriz e a morte celular. Como o MIAT está elevado em várias doenças cardíacas humanas e até circula no sangue como um possível marcador de dano cardíaco, direcionar essa via BACH2–MIAT pode complementar terapias atuais. Estratégias que aumentem a atividade de BACH2, ajustem a sinalização enviesada por β‑arrestina1 com drogas como o carvedilol ou silenciem diretamente o MIAT com ferramentas baseadas em RNA podem, um dia, ajudar a evitar que corações danificados se tornem rígidos e falhem.
Citação: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Palavras-chave: insuficiência cardíaca, fibrose cardíaca, RNA longo não codificante, terapia com beta-bloqueador, apoptose de cardiomiócitos