Clear Sky Science · de
BACH2 verbindet β1‑Adrenerg-Rezeptor/β‑Arrestin1‑Signale mit MIAT, um die Aktivierung kardialer Fibroblasten und Kardiomyozyten-Apoptose zu hemmen
Warum diese Herzstudie wichtig ist
Herzinfarkte und chronische Herzinsuffizienz sind weiterhin führende Todesursachen, teils weil geschädigter Herzmuskel durch Narbengewebe ersetzt wird und lebenswichtige Herzzellen absterben. Diese Studie entdeckt einen zuvor unbekannten Sicherheitsschalter in Herzzellen, der durch ein verbreitetes Betablocker‑Medikament aktiviert wird und danach ein schädliches RNA‑Signal, das mit Vernarbung und Zelltod verbunden ist, ausschaltet. Das Verständnis dieses eingebauten Schutzwegs könnte auf präzisere Therapien hindeuten, die geschädigte Herzen länger und besser arbeiten lassen.
Eine schädliche Botschaft in versagenden Herzen
Wenn ein Teil des Herzens nicht genug Blut bekommt, sterben viele arbeitende Muskelzellen (Kardiomyozyten) und werden durch steife Narben ersetzt, die von Stützzellen namens Fibroblasten gebildet werden. Frühere Forschung identifizierte eine lange nichtkodierende RNA namens MIAT, die wie eine toxische Botschaft wirkt: Sie steigert Gene, die Vernarbung und programmierten Zelltod fördern. MIAT‑Spiegel sind in mehreren menschlichen Herzerkrankungen und in verschiedenen Tiermodellen von Herzverletzung erhöht, während das Blockieren von MIAT bei Mäusen die Herzfunktion nach einem Herzinfarkt verbessert. Bislang war jedoch unklar, was MIAT reguliert und wie Standard‑Herzmedikamente darauf einwirken könnten.
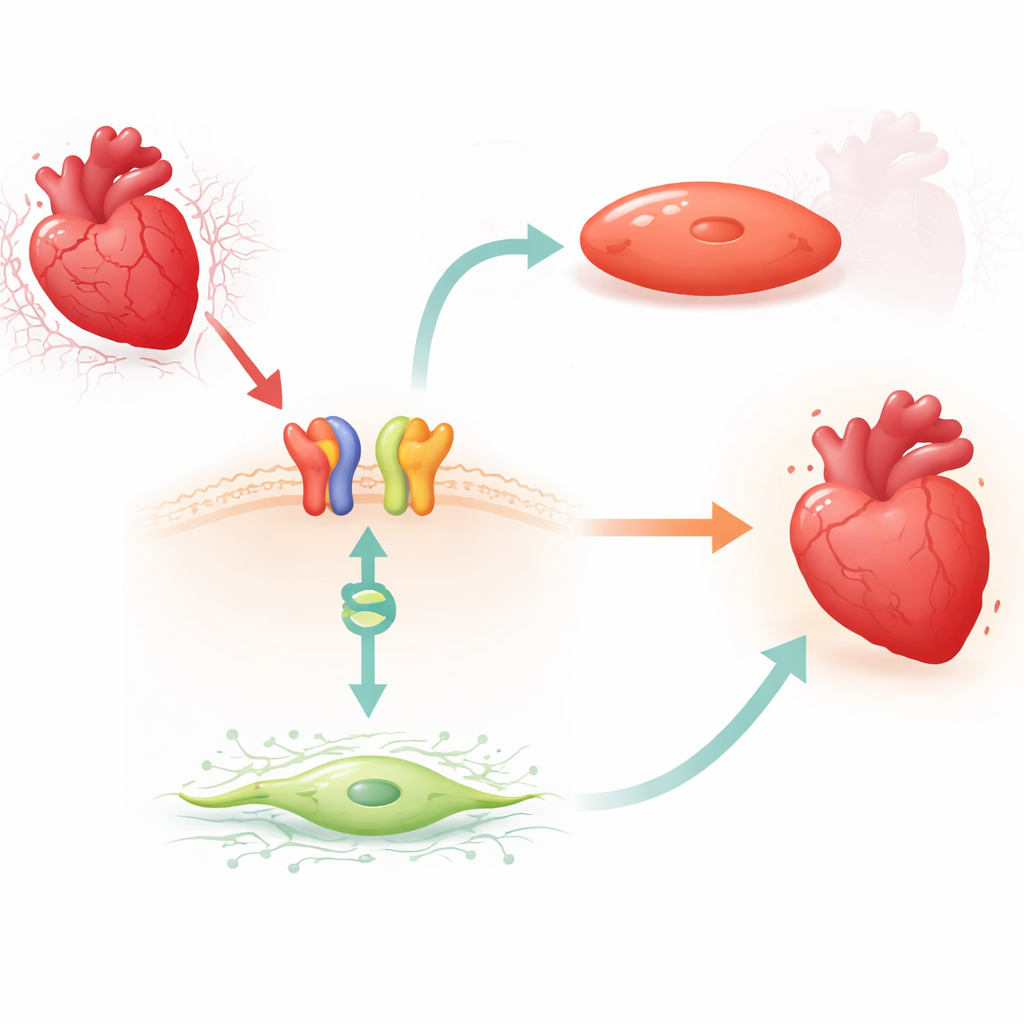
Eine medikamentenaktivierte Verbindung vom Oberflächenschild zur Genumschaltung
Die Forscher konzentrierten sich auf Carvedilol, einen weit verbreiteten Betablocker, der eine schützende Signalkaskade über den β1‑Adrenerg‑Rezeptor und ein Adapterprotein namens β‑Arrestin1 auslösen kann. In Mäusen fanden sie, dass eine kurze Carvedilol‑Behandlung konsistent die MIAT‑Spiegel im linken Ventrikel senkte, jedoch nur, wenn β1‑Rezeptoren und β‑Arrestin1 vorhanden waren. Ähnliche MIAT‑Senkungen traten in humanen adulten kardialen Fibroblasten sowie in menschlichen und tierischen Herzmuskelzellen auf, die in einem Labormodell von Untersauerstoffung und Reoxygenierung exponiert wurden. In all diesen Systemen erhöhte Stress MIAT, während Carvedilol diesen Anstieg umkehrte, was darauf hindeutet, dass das Medikament eine natürliche Bremse gegen die schädliche RNA nutzt.
Das Wächterprotein, das die schädliche RNA beruhigt
Um tiefer zu verstehen, suchte das Team nach einem DNA‑bindenden Protein, das mit β‑Arrestin1 im Zellkern zusammenarbeiten könnte. Sie fokussierten sich auf BACH2, einen Transkriptionsfaktor, der zuvor mit Schutz vor anderen Formen von Herzverletzung in Verbindung gebracht wurde, aber in diesem Kontext noch nicht untersucht war. Mithilfe biochemischer Bindungstests zeigten sie, dass BACH2 an konservierten Abschnitten im MIAT‑Promotor bindet, der Kontrollregion, die die MIAT‑Produktion antreibt. In humanen Herzfibroblasten und Kardiomyozyten reduzierte eine Erhöhung von BACH2 die MIAT‑Spiegel, während das Stilllegen von BACH2 MIAT ansteigen ließ. Wichtig ist, dass Herzen von Patienten mit fortgeschrittener Herzinsuffizienz und von Mäusen nach Herzinfarkt dasselbe Muster zeigten: BACH2‑Spiegel waren niedrig, wenn MIAT hoch war. Carvedilol‑Behandlung bei Mäusen erhöhte die BACH2‑Expression auf β1‑ und β‑Arrestin1‑abhängige Weise und verband damit das äußere Medikamentensignal mit diesem nukleären Genumschalter.
Schutz der Herzzellen vor Vernarbung und Tod
Das Team fragte dann, was BACH2 konkret in unterstützenden und Muskelzellen des Herzens bewirkt. In kultivierten humanen kardialen Fibroblasten förderte der Verlust von BACH2 Fibrose‑Marker, erhöhte Zellwachstum und verstärkte Zellmigration — Verhaltensweisen, die Narbenbildung begünstigen —, während zusätzliches BACH2 diese fibrösen Merkmale abschwächte. In humanen und murinen Kardiomyozyten führte die Reduktion von BACH2 zu mehr abgestorbenen Zellen und höherer Aktivität von zelltod‑assoziierten Enzymen sowie zu verminderten Spiegeln natürlicher Überlebensproteine. Eine Überexpression von BACH2 hatte die entgegengesetzten Effekte und bewahrte das Überleben der Zellen unter Stress. Zusammengenommen stützen diese Experimente ein Modell, in dem Carvedilol β1‑Rezeptoren und β‑Arrestin1 aktiviert, die dann mit BACH2 im Zellkern zusammenarbeiten, um MIAT herunterzufahren und dadurch Vernarbung und Zellverlust zu begrenzen.
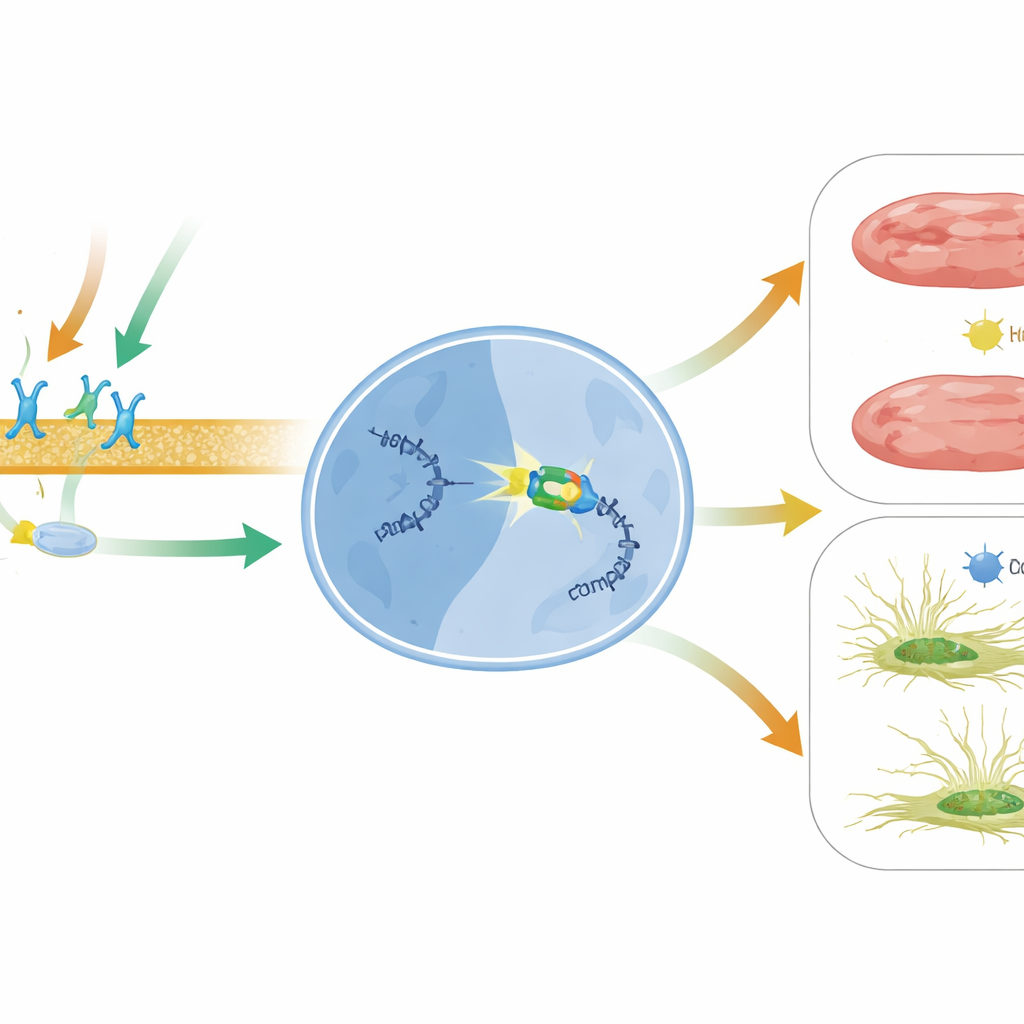
Was dies für zukünftige Herzbehandlungen bedeuten könnte
Vereinfacht gesagt offenbart diese Arbeit eine neue Schutzachse im versagenden Herzen: Medikamenten‑ausgelöste β1‑Rezeptor‑ und β‑Arrestin1‑Signalgebung schaltet BACH2 an, das dann die schädliche MIAT‑Botschaft deaktiviert, welche Vernarbung und Zelltod fördert. Da MIAT in mehreren menschlichen Herzerkrankungen erhöht ist und sogar im Blut als potenzieller Marker für Herzschädigung zirkuliert, könnte das Ansprechen auf den BACH2–MIAT‑Weg aktuelle Therapien ergänzen. Strategien, die BACH2‑Aktivität verstärken, β‑Arrestin1‑bevorzugte Signalgebung mit Wirkstoffen wie Carvedilol feinjustieren oder MIAT direkt mit RNA‑basierten Werkzeugen zum Schweigen bringen, könnten eines Tages helfen, geschädigte Herzen vor Versteifung und Versagen zu bewahren.
Zitation: Moukette, B., Teoh, Jp., Hashmi, W.J. et al. BACH2 links β1-adrenergic receptor/β-arrestin1 signaling to MIAT to inhibit cardiac fibroblast activation and cardiomyocyte apoptosis. Cell Death Discov. 12, 127 (2026). https://doi.org/10.1038/s41420-026-02985-4
Schlüsselwörter: Herzinsuffizienz, kardiale Fibrose, lange nichtkodierende RNA, Betablocker‑Therapie, Kardiomyozyten-Apoptose