Clear Sky Science · pt
Uma interação complexa entre vários motivos intracelulares determina a ligação e ativação de proteínas G pelos receptores muscarínicos
Por que pequenos interruptores celulares importam para a medicina
A cada segundo, nossas células dependem de pequenos interruptores moleculares chamados receptores para detectar hormônios, neurotransmissores e fármacos. Uma classe importante desses interruptores, conhecida como receptores acoplados à proteína G, localiza‑se na membrana celular e transmite mensagens para o interior por meio de parceiros chamados proteínas G. Alguns receptores conversam com apenas um tipo de proteína G, enquanto outros transmitem para vários, influenciando desde a frequência cardíaca até o humor. Este estudo coloca uma pergunta aparentemente simples, com grandes implicações para o desenho de medicamentos: que características dentro de um receptor determinam com quais proteínas G ele se comunica — e com que intensidade?
Torres de celular na membrana
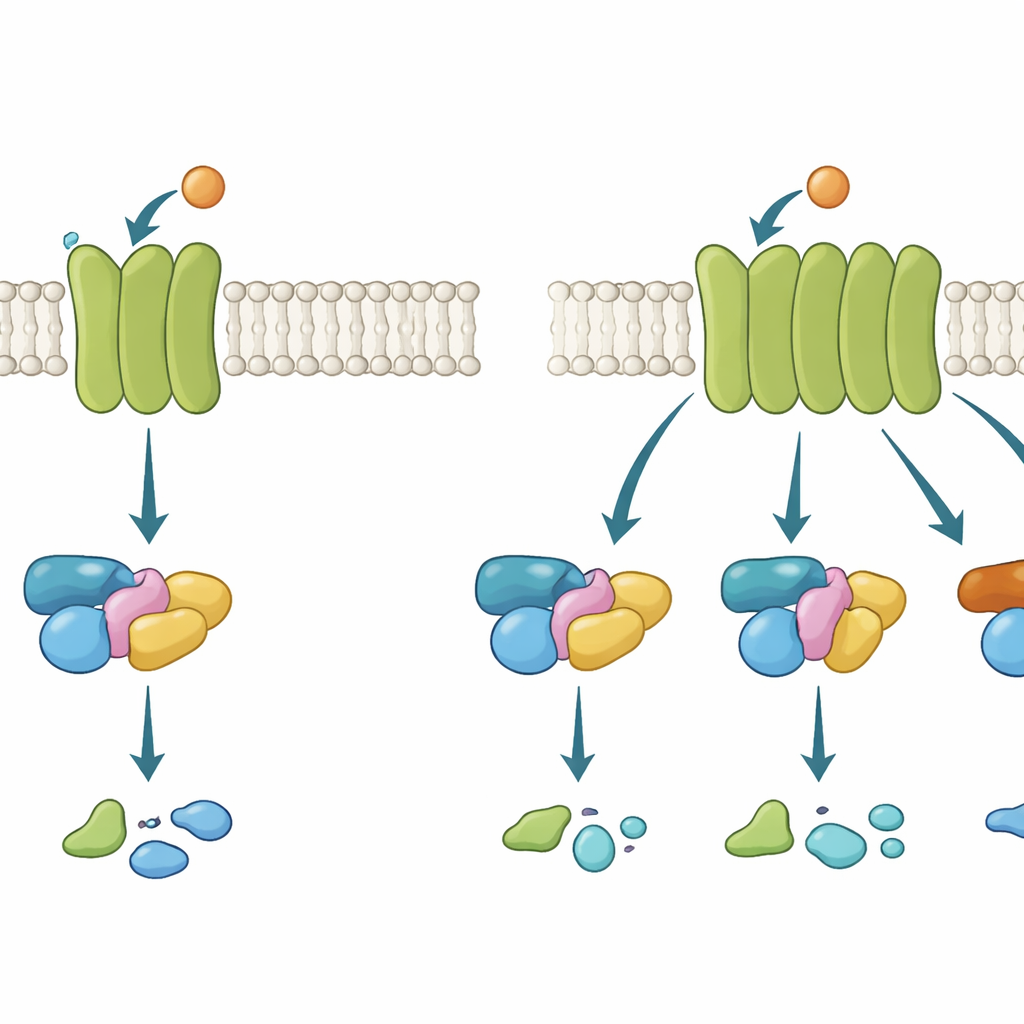
Receptores acoplados à proteína G (GPCRs) funcionam um pouco como torres de celular embutidas na camada externa da célula. Quando uma molécula sinalizadora, como o neurotransmissor acetilcolina, se liga na face externa, o receptor muda de forma na parte interna e recruta uma proteína G. Essa proteína então passa do estado inativo para o ativo e transmite a mensagem adiante. Existem várias famílias de proteínas G, cada uma desencadeando respostas celulares diferentes. Alguns GPCRs são altamente exigentes, usando apenas uma família; outros são mais promiscuos, envolvendo várias. Entender o que torna um receptor seletivo ou flexível é crucial, porque muitos medicamentos de grande impacto atuam ao modular essas vias.
Trocando partes para reescrever o sinal
Os pesquisadores concentraram‑se em dois receptores de acetilcolina intimamente relacionados, um no cérebro e outro no corpo: o receptor M2, que normalmente sinaliza principalmente via uma proteína G do tipo Gi/o, e o receptor M3, que prefere uma proteína G do tipo Gq, mas também pode interagir fracamente com Gi/o. Em vez de buscar um único aminoácido “mágico”, a equipe adotou uma abordagem modular. Construíram receptores quiméricos — híbridos nos quais trocaram segmentos internos inteiros entre M2 e M3, como as pequenas alças intracelulares, as extremidades internas de dois helices-chave (chamados 5 e 6), uma curta hélice de cauda e trechos da cauda ricos em resíduos positivamente carregados. Em seguida perguntaram duas coisas para cada quimera: ela se liga a uma dada proteína G e, em caso afirmativo, consegue ativá‑la com sucesso?
Observando ligação e ativação separadamente
Para rastrear essas interações microscópicas, a equipe usou métodos baseados em luz em células vivas. Em um primeiro conjunto de experimentos, empregaram uma técnica chamada FRET para detectar com que estabilidade um receptor mantém uma proteína G em um ambiente controlado onde o nucleotídeo energético GTP está ausente. Isso revela com que probabilidade e por quanto tempo um par receptor–proteína G permanece unido. Em um segundo conjunto de experimentos, usaram um método relacionado, BRET, com sensores especializados para medir se as proteínas G ligadas efetivamente mudam para seu estado ativo. Ao comparar como os receptores selvagens e quiméricos lidavam com duas famílias de proteínas G — Gi/o e Gq — os pesquisadores puderam separar quais segmentos estruturais favoreciam qual parceiro e em que etapa.
Muitas pequenas mudanças, grandes alterações no comportamento
Os resultados mostraram que nenhuma característica interna única atuava como um interruptor liga/desliga para a escolha da proteína G. Em vez disso, combinações de motivos atuavam em conjunto, e seus efeitos dependiam do contexto do receptor. Por exemplo, transplantar as extremidades internas dos helices 5 e 6 do receptor mais flexível M3 para o seletivo M2 deslocou seu sinalamento para favorecer principalmente Gq em vez de Gi/o, em alguns casos transformando o M2 em um receptor predominantemente acoplado a Gq. Adicionar segmentos adicionais das alças poderia restaurar a capacidade de ligar e ativar ambas as famílias de proteínas G. Inversamente, inserir segmentos no estilo M2 no M3 podia fazê‑lo favorecer Gi/o com mais força ou, com alterações na sua cauda, empurrá‑lo para sinalizar quase exclusivamente via Gq. Surpreendentemente, algumas quimeras podiam ligar uma proteína G sem ativá‑la, enquanto outras ainda conseguiam disparar ativação mesmo quando a ligação era tão fugaz que não era detectável pelo ensaio de ligação.
O que isso significa para futuros medicamentos
Em termos práticos, este trabalho mostra que os GPCRs escolhem seus parceiros de sinalização não por uma única fechadura, mas por toda uma combinação de partes internas. As alças internas, as extremidades internas de determinados helices e a cauda contribuem de forma coordenada para determinar se um receptor prefere uma família de proteína G, várias, ou pode se ligar sem realmente ativá‑las. Ao reorganizar habilidosamente esses segmentos, os autores conseguiram reprogramar receptores de seletivos para amplamente sinalizantes e vice‑versa. Para a descoberta de fármacos, isso sugere que ajustar como um receptor usa seus vários “botões” internos poderia permitir que medicamentos futuros direcionassem sinais para vias benéficas e afastassem efeitos colaterais, sem a necessidade de redesenhar completamente o receptor em si.
Citação: Kirchhofer, S.B., Jelinek, V., Klingelhöfer, K. et al. A complex interplay of various intracellular motifs determines G protein binding and activation of muscarinic receptors. Sci Rep 16, 12370 (2026). https://doi.org/10.1038/s41598-026-48667-0
Palavras-chave: receptores acoplados à proteína G, receptores muscarínicos, transdução de sinal, seletividade do receptor, alvo farmacológico