Clear Sky Science · de
Ein komplexes Zusammenspiel verschiedener intrazellulärer Motive bestimmt die G‑Protein‑Bindung und Aktivierung muscarinerger Rezeptoren
Warum winzige Zellschalter für die Medizin wichtig sind
Sekündlich verlassen sich unsere Zellen auf winzige molekulare Schalter, sogenannte Rezeptoren, um Hormone, Neurotransmitter und Medikamente zu erkennen. Eine große Klasse dieser Schalter, bekannt als G‑Protein‑gekoppelte Rezeptoren, sitzt in der Zellmembran und übermittelt Botschaften ins Zellinnere über Partner namens G‑Proteine. Manche Rezeptoren kommunizieren nur mit einem G‑Protein‑Typ, andere senden an mehrere und beeinflussen so alles von Herzfrequenz bis Stimmung. Diese Studie stellt eine auf den ersten Blick einfache, aber für die Wirkstoffentwicklung bedeutsame Frage: Welche Merkmale innerhalb eines Rezeptors entscheiden, mit welchen G‑Proteinen er kommuniziert — und wie stark?
Funkmasten auf der Membran
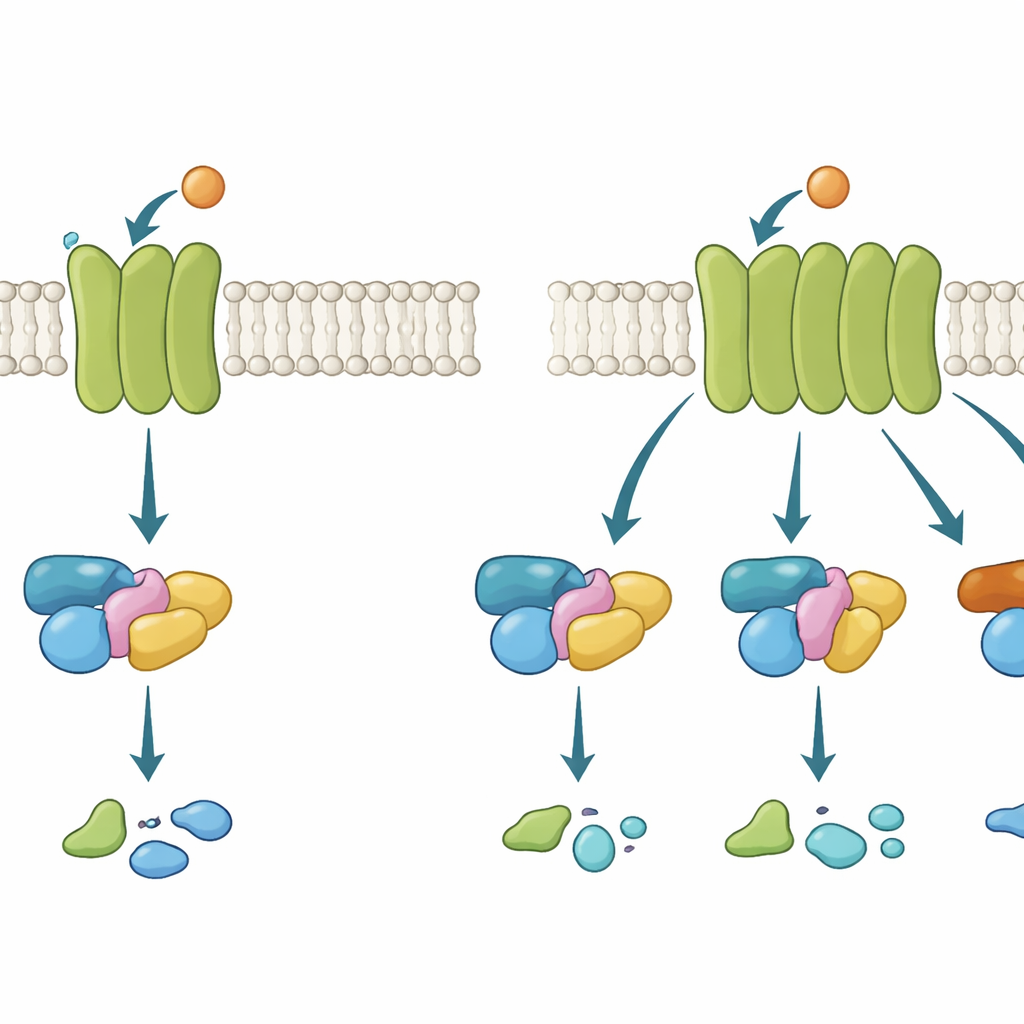
G‑Protein‑gekoppelte Rezeptoren (GPCRs) funktionieren ein wenig wie Funkmasten in der Hülle der Zelle. Wenn ein Signalstoff, etwa der Neurotransmitter Acetylcholin, außen bindet, ändert der Rezeptor innen seine Konformation und rekrutiert ein G‑Protein. Dieses wechselt dann vom inaktiven in den aktiven Zustand und leitet die Nachricht weiter. Es gibt mehrere G‑Protein‑Familien, die unterschiedliche zelluläre Antworten auslösen. Manche GPCRs sind sehr wählerisch und nutzen nur eine Familie; andere sind flexibler und binden mehrere. Zu verstehen, was einen Rezeptor selektiv oder flexibel macht, ist entscheidend, weil viele Blockbuster‑Medikamente diese Wege beeinflussen.
Teile tauschen, Signal neu schreiben
Die Forscher konzentrierten sich auf zwei eng verwandte Acetylcholinrezeptoren im Gehirn und Körper: den M2‑Rezeptor, der normalerweise vorwiegend über ein Gi/o‑Typ‑G‑Protein signalisiert, und den M3‑Rezeptor, der Gq‑Typ‑G‑Proteine bevorzugt, aber auch schwach Gi/o binden kann. Statt nach einzelnen „magischen“ Aminosäuren zu suchen, verfolgte das Team einen modularen Ansatz. Sie konstruierten chimäre Rezeptoren — Hybride, bei denen sie ganze intrazelluläre Segmente zwischen M2 und M3 austauschten, etwa kurze intrazelluläre Schleifen, die inneren Enden zweier wichtiger Helices (genannt 5 und 6), eine kurze Schwanzhelix und Abschnitte der C‑Terminal‑Region mit vielen positiv geladenen Resten. Für jede Chimäre stellten sie zwei getrennte Fragen: Bindet sie ein gegebenes G‑Protein, und wenn ja, aktiviert sie es auch erfolgreich?
Bindung und Aktivierung getrennt beobachten
Um diese mikroskopischen Wechselwirkungen zu verfolgen, nutzte das Team lichtbasierte Methoden in lebenden Zellen. In einem ersten Experiment setzten sie eine Technik namens FRET ein, um zu detektieren, wie stabil ein Rezeptor ein G‑Protein in einem kontrollierten Setting hält, in dem das übliche energietragende Nukleotid GTP fehlt. Das zeigt, wie wahrscheinlich und wie lang ein Rezeptor–G‑Protein‑Paar zusammenbleibt. In einer zweiten Experimentreihe verwendeten sie eine verwandte Methode, BRET, mit spezialisierten Sensorconstructs, um zu messen, ob die gebundenen G‑Proteine tatsächlich in ihren aktiven Zustand übergehen. Durch den Vergleich, wie Wildtyp‑ und chimäre Rezeptoren mit zwei G‑Protein‑Familien — Gi/o und Gq — umgingen, konnten die Forscher herausarbeiten, welche strukturellen Segmente welchen Partner in welchem Schritt begünstigten.
Viele kleine Änderungen, große Verhaltensverschiebungen
Die Ergebnisse zeigten, dass kein einzelnes internes Merkmal als Ein‑/Ausschalter für die G‑Protein‑Wahl fungiert. Stattdessen wirkten Kombinationen von Motiven zusammen, und ihre Effekte hingen vom Rezeptorhintergrund ab. Beispielsweise verschob das Einpflanzen der inneren Enden der Helices 5 und 6 aus dem flexibleren M3 in den selektiven M2 diesen hin zu einer vorwiegenden Gq‑Signalisierung statt Gi/o und machte M2 in einigen Fällen zu einem überwiegend Gq‑gekoppelten Rezeptor. Das Hinzufügen weiterer Schleifenabschnitte konnte die Fähigkeit wiederherstellen, beide G‑Protein‑Familien zu binden und zu aktivieren. Umgekehrt konnte das Einfügen M2‑ähnlicher Segmente in M3 dazu führen, dass dieser stärker Gi/o bevorzugte oder mit Änderungen im Schwanz fast ausschließlich über Gq signalisierte. Auffällig war, dass einige Chimären ein G‑Protein binden konnten, ohne es zu aktivieren, während andere noch aktivieren konnten, obwohl die Bindung zu kurzlebig war, um im Bindungsassay nachgewiesen zu werden.
Was das für künftige Medikamente bedeutet
Anschaulich zeigt diese Arbeit, dass GPCRs ihre Signalpartner nicht durch ein einzelnes Schlüsselloch wählen, sondern durch ein ganzes Zahlenschloss aus inneren Teilen. Die inneren Schleifen, die inneren Enden bestimmter Helices und der Schwanz tragen koordiniert dazu bei, ob ein Rezeptor eine G‑Protein‑Familie, mehrere Familien bevorzugt oder binden kann, ohne sie wirklich zu aktivieren. Durch geschicktes Umordnen dieser Segmente konnten die Autoren Rezeptoren von selektiv zu breit signalisierend und wieder zurückprogrammieren. Für die Wirkstoffforschung deutet das darauf hin, dass das Feinjustieren, wie ein Rezeptor seine verschiedenen inneren „Drehknöpfe“ nutzt, es künftigen Medikamenten erlauben könnte, Signale in nützliche Bahnen zu lenken und Nebenwirkungen zu vermeiden, ohne den Rezeptor komplett neu entwerfen zu müssen.
Zitation: Kirchhofer, S.B., Jelinek, V., Klingelhöfer, K. et al. A complex interplay of various intracellular motifs determines G protein binding and activation of muscarinic receptors. Sci Rep 16, 12370 (2026). https://doi.org/10.1038/s41598-026-48667-0
Schlüsselwörter: G‑Protein‑gekoppelte Rezeptoren, muscarinerge Rezeptoren, Signaltransduktion, Rezeptorselektivität, Wirkstoffzielausrichtung