Clear Sky Science · es
Una interacción compleja de varios motivos intracelulares determina la unión y activación de proteínas G en los receptores muscarínicos
Por qué importan a la medicina estos pequeños interruptores celulares
Cada segundo, nuestras células dependen de diminutos interruptores moleculares llamados receptores para detectar hormonas, neurotransmisores y fármacos. Una clase importante de estos interruptores, conocida como receptores acoplados a proteínas G, se sitúa en la membrana celular y transmite mensajes al interior mediante socios llamados proteínas G. Algunos receptores se comunican con un solo tipo de proteína G, mientras que otros emiten señales a varios tipos, modulando desde la frecuencia cardíaca hasta el estado de ánimo. Este estudio plantea una pregunta aparentemente simple con grandes implicaciones para el diseño de fármacos: ¿qué rasgos dentro de un receptor determinan con qué proteínas G se comunica—y con qué intensidad?
Torres de telefonía móvil en la membrana
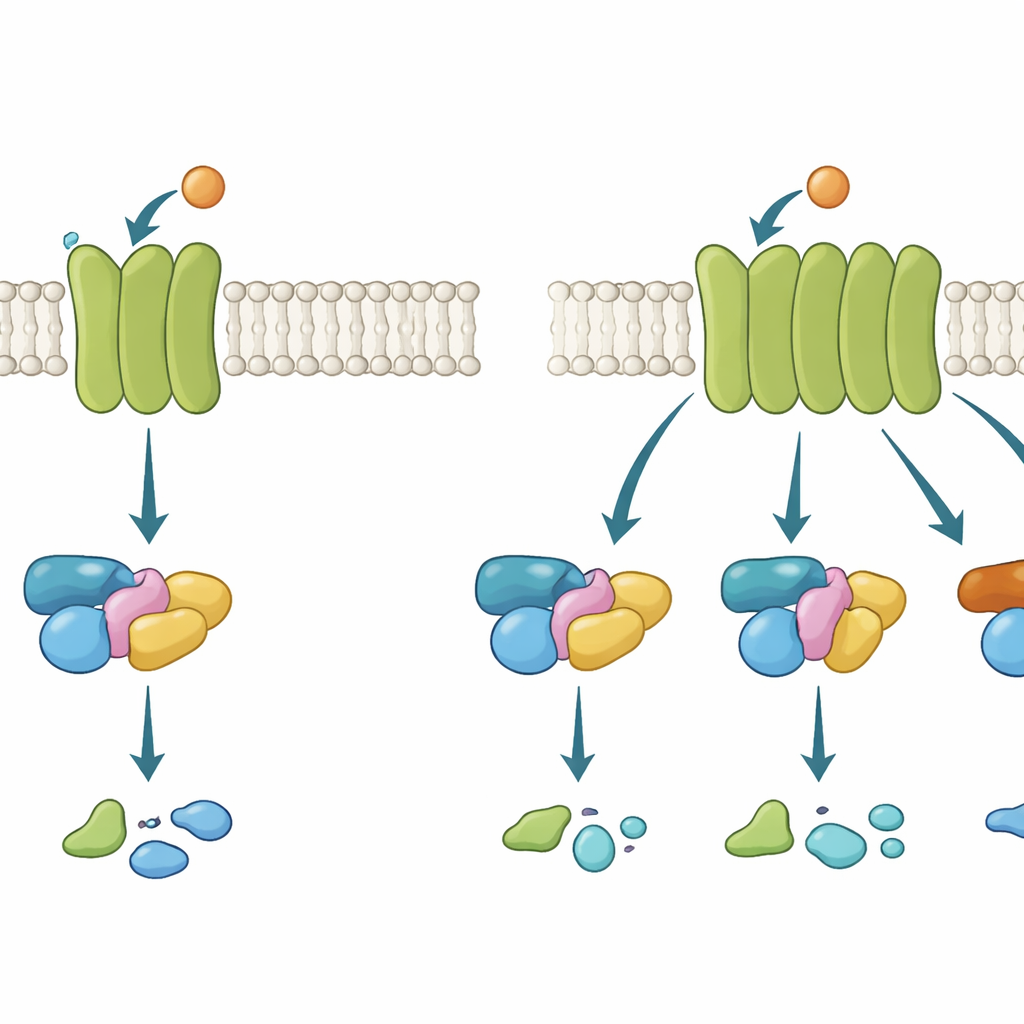
Los receptores acoplados a proteínas G (GPCR) actúan un poco como torres de telefonía móvil incrustadas en la envoltura exterior de la célula. Cuando una molécula señal, como el neurotransmisor acetilcolina, se une en el exterior, el receptor cambia de forma en el interior y recluta una proteína G. Esa proteína G pasa entonces de un estado inactivo a uno activo y transmite el mensaje. Existen varias familias de proteínas G, cada una desencadenando respuestas celulares distintas. Algunos GPCR son muy selectivos y usan solo una familia; otros son más promiscuos y comprometen varias. Entender qué hace que un receptor sea selectivo o flexible es crucial, porque muchos fármacos de gran impacto funcionan modulando estas vías.
Intercambiar partes para reescribir la señal
Los investigadores se centraron en dos receptores de acetilcolina estrechamente relacionados en el cerebro y el cuerpo: el receptor M2, que normalmente señaliza principalmente a través de una proteína G tipo Gi/o, y el receptor M3, que prefiere una proteína G tipo Gq pero que también puede comprometer débilmente a Gi/o. En lugar de buscar un único aminoácido “mágico”, el equipo adoptó un enfoque modular. Construyeron receptores quiméricos—híbridos en los que intercambiaron segmentos internos enteros entre M2 y M3, tales como las cortas asas dentro de la célula, los extremos internos de dos hélices clave (denominadas 5 y 6), una corta hélice de cola y tramos de la cola ricos en residuos cargados positivamente. Luego formularon dos preguntas separadas para cada quimera: ¿se une a una proteína G dada y, de ser así, la activa con éxito?
Observar por separado la unión y la activación
Para seguir estas interacciones microscópicas, el equipo empleó métodos basados en luz en células vivas. En un primer conjunto de experimentos usaron una técnica llamada FRET para detectar cuán establemente un receptor se mantiene unido a una proteína G en un entorno controlado donde falta el nucleótido energizante habitual, el GTP. Esto revela con qué probabilidad y durante cuánto tiempo un par receptor–proteína G permanece unido. En un segundo conjunto de experimentos utilizaron un método relacionado, BRET, con constructos sensores especializados para medir si las proteínas G enlazadas efectivamente pasan a su estado activo. Al comparar cómo los receptores silvestres y quiméricos manejaban dos familias de proteínas G—Gi/o y Gq—los investigadores pudieron desentrañar qué segmentos estructurales favorecían a cada socio y en qué etapa.
Muchos cambios pequeños, grandes desplazamientos en el comportamiento
Los resultados mostraron que ninguna característica interna individual actuaba como un interruptor encendido–apagado para la elección de la proteína G. En cambio, combinaciones de motivos actuaban conjuntamente y sus efectos dependían del contexto del receptor. Por ejemplo, trasplantar los extremos internos de las hélices 5 y 6 del más flexible receptor M3 al receptor selectivo M2 lo desplazó a señalizar principalmente a través de Gq en lugar de Gi/o, en algunos casos convirtiendo a M2 en un receptor acoplado predominantemente a Gq. Añadir además segmentos de las asas podía restaurar la capacidad de unirse y activar a ambas familias de proteínas G. Por el contrario, insertar segmentos tipo M2 en M3 podía hacer que este favoreciera más fuertemente a Gi/o o, con cambios en su cola, empujarlo hacia una señalización casi exclusiva por Gq. Llamativamente, algunas quimeras pudieron unirse a una proteína G sin activarla, mientras que otras aún podían desencadenar la activación incluso cuando la unión era demasiado fugaz para detectarse con el ensayo de unión.
Qué significa esto para futuros fármacos
En términos cotidianos, este trabajo muestra que los GPCR eligen a sus socios de señalización no mediante una única cerradura, sino a través de una combinación de partes internas. Las asas internas, los extremos internos de ciertas hélices y la cola contribuyen de forma coordinada a si un receptor prefiere una familia de proteínas G, varias, o puede unirse sin realmente activarlas. Al reorganizar hábilmente estos segmentos, los autores pudieron reprogramar receptores de selectivos a de señalización amplia y viceversa. Para el descubrimiento de fármacos, esto sugiere que ajustar cómo un receptor emplea sus distintos “mandos” internos podría permitir que medicinas futuras dirijan las señales hacia vías beneficiosas y alejen los efectos secundarios, sin necesidad de rediseñar completamente el receptor en sí.
Cita: Kirchhofer, S.B., Jelinek, V., Klingelhöfer, K. et al. A complex interplay of various intracellular motifs determines G protein binding and activation of muscarinic receptors. Sci Rep 16, 12370 (2026). https://doi.org/10.1038/s41598-026-48667-0
Palabras clave: Receptores acoplados a proteínas G, receptores muscarínicos, transducción de señales, selectividad de receptores, dirigido de fármacos