Clear Sky Science · pt
Otimização e pesquisa comparativa do modelo BERT-SPAN com modularização adaptativa para reconhecimento de entidades nomeadas de proteínas do vírus SARS-CoV-2
Por que encontrar nomes de proteínas em artigos é importante
Cada semana surgem milhares de novos artigos científicos sobre COVID-19 e seu vírus, o SARS-CoV-2. Escondidas nesses textos densos estão pistas cruciais sobre as proteínas do vírus — máquinas moleculares que o ajudam a infectar nossas células e que são alvos primários para medicamentos e vacinas. Mas nenhum humano consegue ler tudo. Este estudo trata de uma questão simples, porém poderosa: podemos ensinar computadores a identificar automaticamente e listar as proteínas do vírus em texto, com rapidez e precisão suficientes para ajudar pesquisadores a acompanhar o volume?
Transformando texto congestionado em dados utilizáveis
Quando cientistas escrevem sobre o SARS-CoV-2, usam uma mistura confusa de nomes completos, abreviações e apelidos para suas proteínas. A mesma proteína pode aparecer como “spike protein”, “S” ou dentro de frases como “domínio de ligação ao receptor da spike”. Para entender essa linguagem em grande escala, os computadores precisam primeiro de um “corpus” — uma coleção cuidadosamente rotulada de frases onde especialistas destacaram cada menção a proteína. Os autores construíram esse recurso ao filtrar cerca de 200.000 resumos de coronavírus do PubMed e então rotular manualmente todas as menções a proteínas do SARS-CoV-2 nas frases relevantes. Após limpeza e checagens cruzadas, eles ficaram com 7.159 frases e 12.660 menções de proteínas rotuladas que servem como verdade de referência para treinar e testar algoritmos.
Ensinando máquinas a ler nomes de proteínas virais
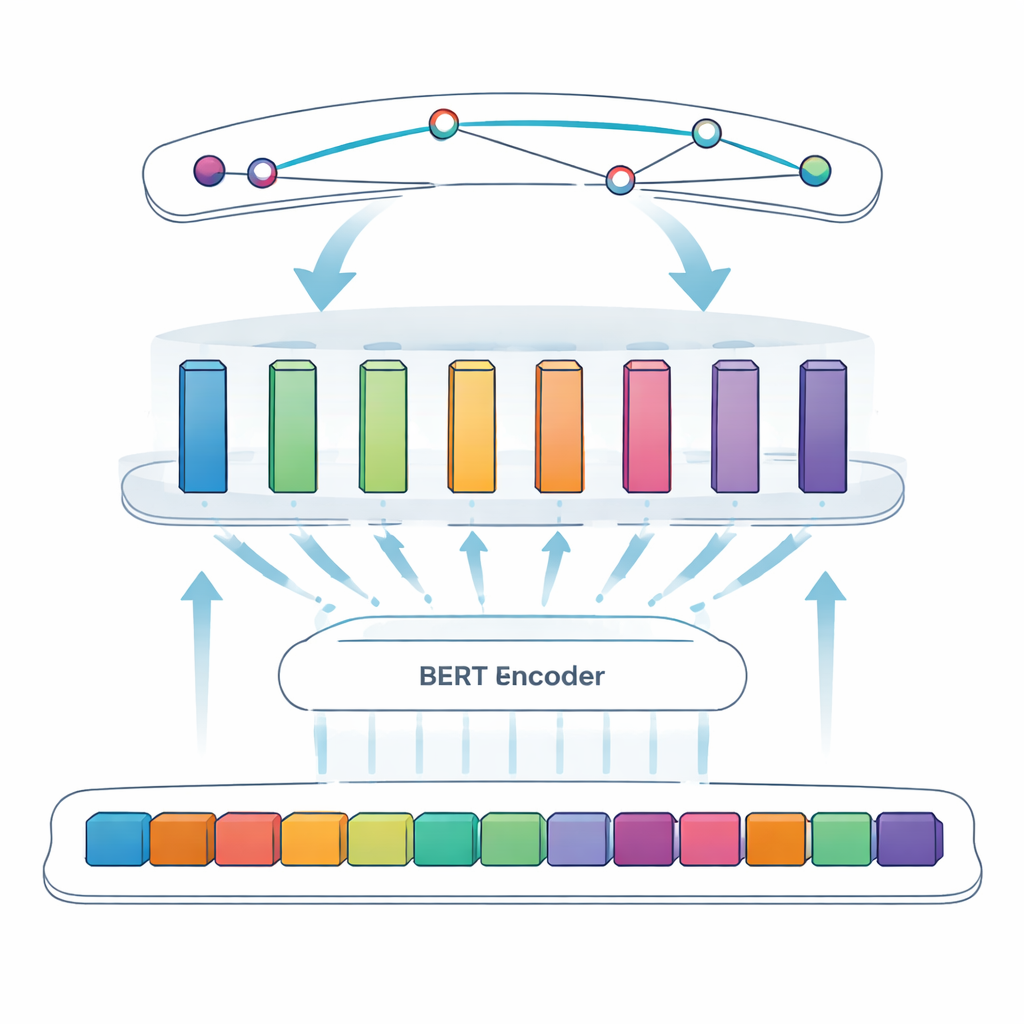
A tarefa central deste estudo é chamada reconhecimento de entidades nomeadas: fazer um computador identificar itens específicos — neste caso, proteínas virais — em texto livre. Abordagens anteriores dependiam de regras manuais ou listas de termos conhecidos, que falham quando a nomenclatura é irregular ou evolui. Sistemas mais novos usam aprendizado profundo, em que um modelo aprende padrões diretamente a partir de exemplos. A equipe parte do BERT, um modelo de linguagem que lê o texto em ambas as direções e aprende representações ricas das palavras com base no contexto. Sobre o BERT, eles usam uma abordagem “SPAN”: em vez de decidir, token por token, se cada unidade faz parte de uma entidade, o modelo prevê diretamente as posições de início e fim de cada menção de proteína. Isso facilita lidar com casos complicados, como nomes aninhados ou de múltiplas palavras.
Acrescentando blocos construtivos inteligentes ao modelo
Para afinar ainda mais o sistema, os autores introduzem uma camada feedforward modular adaptativa, que pode ser vista como vários pequenos módulos especialistas entre o BERT e o detector final de spans. Diferentes módulos se especializam em distintos tipos de padrões — por exemplo, características de superfície como hífens e misturas de letras e números, ou pistas contextuais sutis que indicam quando uma letra curta como “S” é realmente o nome de uma proteína. Um mecanismo de ponderação adaptativa então decide, para cada frase, em quais módulos confiar mais, combinando suas saídas em um sinal global mais forte antes da etapa de predição de spans. Esse desenho permite ao modelo focar de forma flexível nos indícios mais informativos para cada trecho de texto específico.
Colocando a abordagem à prova
Os pesquisadores compararam rigorosamente seu modelo BERT–SPAN modular adaptativo com vários sistemas estabelecidos, incluindo um popular modelo BiLSTM–CRF e uma versão padrão do BERT–SPAN sem aprimoramentos modulares. Usando medidas padrão de precisão, eles constataram que o tradicional BiLSTM–CRF alcançou uma pontuação F1 (equilíbrio entre precisão e recall) de cerca de 80,8% em seu conjunto de dados de proteínas do SARS-CoV-2. A adoção do BERT–SPAN sozinho elevou o desempenho para 91,85%. A adição da camada modular adaptativa impulsionou a F1 para 93,66%, um ganho significativo em um campo já maduro. Experimentos de ablação cuidadosos — removendo partes da arquitetura uma a uma — mostraram que tanto a modularização quanto a ponderação adaptativa contribuíram para a melhora, especialmente ao reduzir erros de delimitação e confusões entre abreviações semelhantes.
Como isso ajuda pesquisas futuras
Para um leitor não especializado, o resultado chave é que os autores construíram tanto um conjunto de dados de alta qualidade quanto um motor de leitura mais inteligente que pode reconhecer de forma confiável proteínas do SARS-CoV-2 em textos científicos. Ao transformar linguagem desordenada em listas estruturadas de entidades, o sistema facilita muito a construção de grafos de conhecimento, o mapeamento de quais proteínas virais interagem com quais moléculas hospedeiras e o acompanhamento de como a evidência sobre alvos específicos está se acumulando. Na prática, isso significa que computadores podem assumir mais do trabalho tedioso de mineração de texto, liberando especialistas humanos para interpretar resultados e planejar experimentos. O estudo mostra que combinar modelos de linguagem poderosos com componentes modulares e adaptativos é uma receita promissora para peneirar o dilúvio da literatura biomédica — não apenas para a pesquisa sobre coronavírus, mas para muitos desafios futuros em que a ciência supera nossa capacidade de leitura.
Citação: Shi, X., Fei, Z., Nkhata, A. et al. Optimization and comparison research on adaptive modularization BERT-SPAN model for named entity recognition of SARS-CoV-2 virus proteins. Sci Rep 16, 14207 (2026). https://doi.org/10.1038/s41598-026-44317-7
Palavras-chave: proteínas do SARS-CoV-2, reconhecimento de entidades nomeadas, mineração de texto biomédico, modelo de linguagem BERT, grafos de conhecimento