Clear Sky Science · fr
Recherche d'optimisation et de comparaison sur le modèle BERT-SPAN à modularisation adaptative pour la reconnaissance d'entités nommées des protéines du SARS-CoV-2
Pourquoi identifier les noms de protéines dans les articles est important
Toutes les semaines, des milliers de nouveaux articles scientifiques sur la COVID-19 et son virus, le SARS-CoV-2, paraissent en ligne. Cachés dans ces textes denses se trouvent des informations cruciales sur les protéines du virus — des machines moléculaires qui lui permettent d’infecter nos cellules et qui constituent des cibles privilégiées pour des médicaments et des vaccins. Mais aucun humain ne peut tout lire. Cette étude traite une question simple mais puissante : peut-on apprendre aux ordinateurs à repérer automatiquement et à dresser la liste des protéines virales dans le texte, assez rapidement et avec suffisamment de précision pour aider les chercheurs à suivre le rythme ?
Transformer un texte touffu en données exploitables
Quand les scientifiques écrivent sur le SARS-CoV-2, ils utilisent un mélange déroutant de noms complets, d’abréviations et de surnoms pour désigner ses protéines. La même protéine peut apparaître comme « spike protein », « S », ou être noyée dans des expressions telles que « receptor-binding domain of spike ». Pour comprendre ce langage à grande échelle, les ordinateurs ont d’abord besoin d’un corpus — une collection soigneusement annotée de phrases où des experts ont repéré chaque mention de protéine. Les auteurs ont construit une telle ressource en dépouillant environ 200 000 résumés de coronavirus issus de PubMed, puis en annotant manuellement chaque mention de protéine SARS-CoV-2 dans les phrases pertinentes. Après nettoyage et vérifications croisées, ils ont obtenu 7 159 phrases et 12 660 mentions de protéines annotées qui servent de vérité terrain pour entraîner et tester les algorithmes.
Apprendre aux machines à lire les noms de protéines virales
La tâche centrale de cette étude est la reconnaissance d’entités nommées : amener un ordinateur à repérer des éléments spécifiques — ici, des protéines virales — dans un texte libre. Les approches antérieures reposaient sur des règles manuelles ou des lexiques, qui s’effondrent quand la dénomination est irrégulière ou évolutive. Les systèmes plus récents utilisent l’apprentissage profond, où le modèle apprend les motifs directement à partir d’exemples. L’équipe s’appuie sur BERT, un modèle de langage qui lit le texte dans les deux sens et apprend des représentations riches des mots selon leur contexte. Au-dessus de BERT, ils utilisent une approche « SPAN » : au lieu de décider, token par token, si chaque unité appartient à une entité, le modèle prédit directement les positions de début et de fin de chaque mention de protéine. Cela facilite le traitement de cas délicats comme les noms imbriqués ou multi-mots.
Ajouter des blocs modulaires intelligents au modèle
Pour affiner encore le système, les auteurs introduisent une couche feedforward modulaire adaptative, que l’on peut voir comme plusieurs petits modules experts intercalés entre BERT et le détecteur de spans final. Différents modules se spécialisent dans différents types de motifs — par exemple des caractéristiques de surface comme les traits d’union et les mélanges lettres-chiffres, ou des indices contextuels subtils qui signalent quand une courte lettre comme « S » est en réalité un nom de protéine. Un mécanisme de pondération adaptative décide ensuite, pour chaque phrase, quels modules privilégier, en combinant leurs sorties en un signal global plus robuste avant l’étape de prédiction des spans. Cette architecture permet au modèle de se concentrer de manière flexible sur les indices les plus informatifs pour chaque segment de texte.
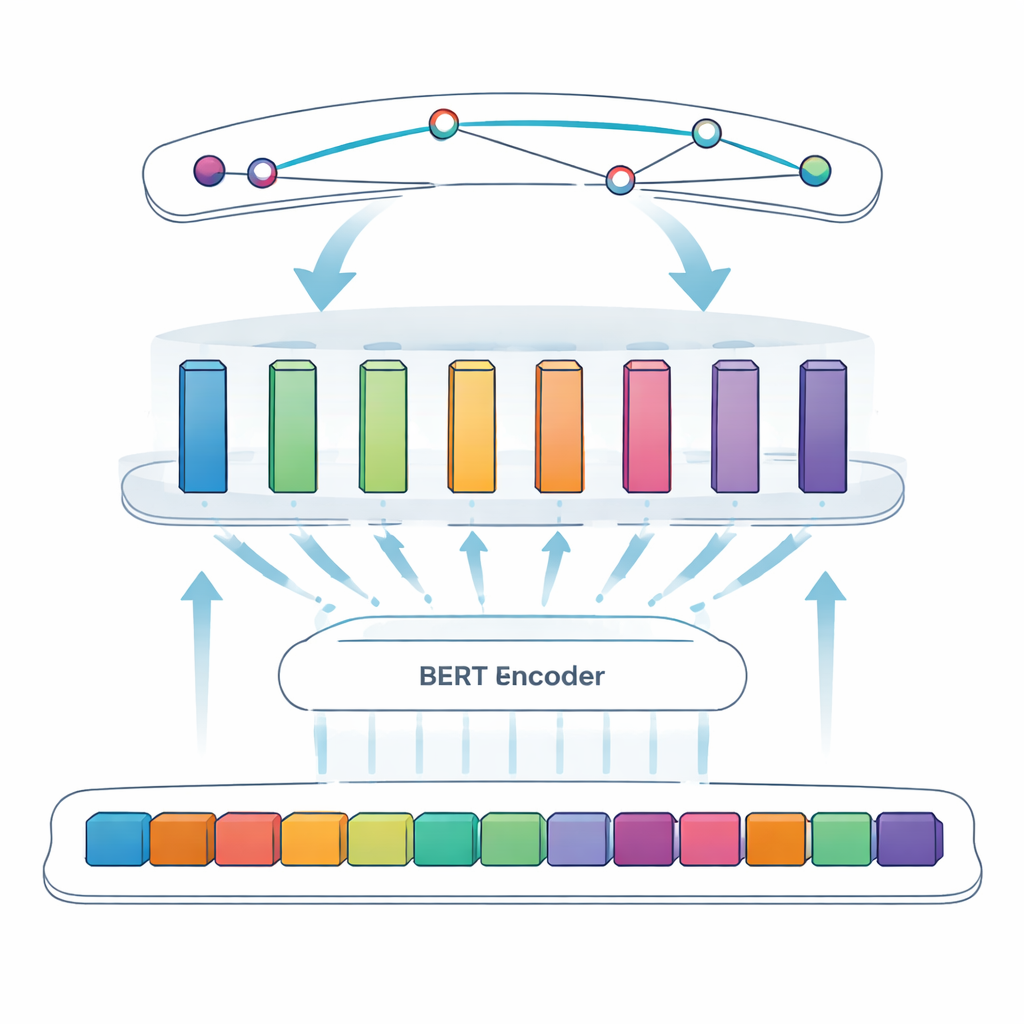
Évaluer l’approche
Les chercheurs ont comparé rigoureusement leur modèle BERT–SPAN modulaire adaptatif à plusieurs systèmes établis, y compris un modèle BiLSTM–CRF populaire et une version standard de BERT–SPAN sans améliorations modulaires. En utilisant des mesures standard d’exactitude, ils ont constaté que le modèle BiLSTM–CRF traditionnel atteignait un score F1 (équilibre entre précision et rappel) d’environ 80,8 % sur leur jeu de données de protéines SARS-CoV-2. Le passage à BERT–SPAN seul a porté la performance à 91,85 %. L’ajout de la couche modulaire adaptative a poussé le score F1 à 93,66 %, un gain significatif dans un domaine déjà mature. Des expériences d’« ablation » — supprimant des composantes de l’architecture une par une — ont montré que la modularisation et la pondération adaptative contribuaient toutes deux à l’amélioration, en réduisant notamment les erreurs de délimitation et les confusions entre abréviations similaires.
Comment cela aide la recherche à venir
Pour le lecteur non spécialiste, le résultat clé est que les auteurs ont construit à la fois un jeu de données de haute qualité et un moteur de lecture plus intelligent capable de reconnaître de manière fiable les protéines du SARS-CoV-2 dans la littérature scientifique. En transformant un langage désordonné en listes structurées d’entités, leur système facilite grandement la construction de graphes de connaissances, la cartographie des interactions entre protéines virales et molécules hôtes, et le suivi de l’accumulation des preuves concernant des cibles particulières. Concrètement, cela signifie que les ordinateurs peuvent prendre en charge une partie plus importante du travail fastidieux d’extraction d’informations textuelles, libérant les experts humains pour interpréter les résultats et concevoir des expériences. L’étude montre qu’associer des modèles de langage puissants à des composants modulaires et adaptatifs est une recette prometteuse pour trier le déluge de littérature biomédicale — non seulement pour la recherche sur le coronavirus, mais pour de nombreux défis futurs où la science dépasse notre capacité de lecture.
Citation: Shi, X., Fei, Z., Nkhata, A. et al. Optimization and comparison research on adaptive modularization BERT-SPAN model for named entity recognition of SARS-CoV-2 virus proteins. Sci Rep 16, 14207 (2026). https://doi.org/10.1038/s41598-026-44317-7
Mots-clés: Protéines du SARS-CoV-2, reconnaissance d'entités nommées, extraction de texte biomédical, modèle de langage BERT, graphes de connaissances