Clear Sky Science · es
Investigación de optimización y comparación del modelo BERT-SPAN con modularización adaptativa para el reconocimiento de entidades nombradas de proteínas del virus SARS-CoV-2
Por qué importa encontrar nombres de proteínas en los artículos
Cada semana aparecen en línea miles de nuevos artículos científicos sobre la COVID-19 y su virus, SARS-CoV-2. Ocultas en esos textos densos hay pistas cruciales sobre las proteínas del virus: máquinas moleculares que le ayudan a infectar nuestras células y que son objetivos principales para fármacos y vacunas. Pero ningún humano puede leerlo todo. Este estudio aborda una pregunta sencilla pero poderosa: ¿podemos enseñar a los ordenadores a detectar y listar automáticamente las proteínas del virus en el texto, con la rapidez y precisión necesarias para ayudar a los investigadores a mantenerse al día?
Convertir texto saturado en datos útiles
Cuando los científicos escriben sobre SARS-CoV-2 usan una mezcla confusa de nombres completos, abreviaturas y apodos para sus proteínas. La misma proteína puede aparecer como “spike protein”, “S” o camuflada en frases como “dominio de unión al receptor de la espiga”. Para entender ese lenguaje a gran escala, los ordenadores necesitan primero un “corpus”: una colección de oraciones cuidadosamente etiquetadas donde expertos han marcado cada mención de proteína. Los autores crearon ese recurso filtrando alrededor de 200.000 resúmenes de coronavirus de PubMed y luego anotando manualmente cada mención de proteínas de SARS-CoV-2 en las oraciones relevantes. Tras depurar y verificar cruzadamente, acabaron con 7.159 oraciones y 12.660 menciones de proteínas etiquetadas que sirven como verdad de referencia para entrenar y evaluar algoritmos.
Enseñar a las máquinas a leer nombres de proteínas virales
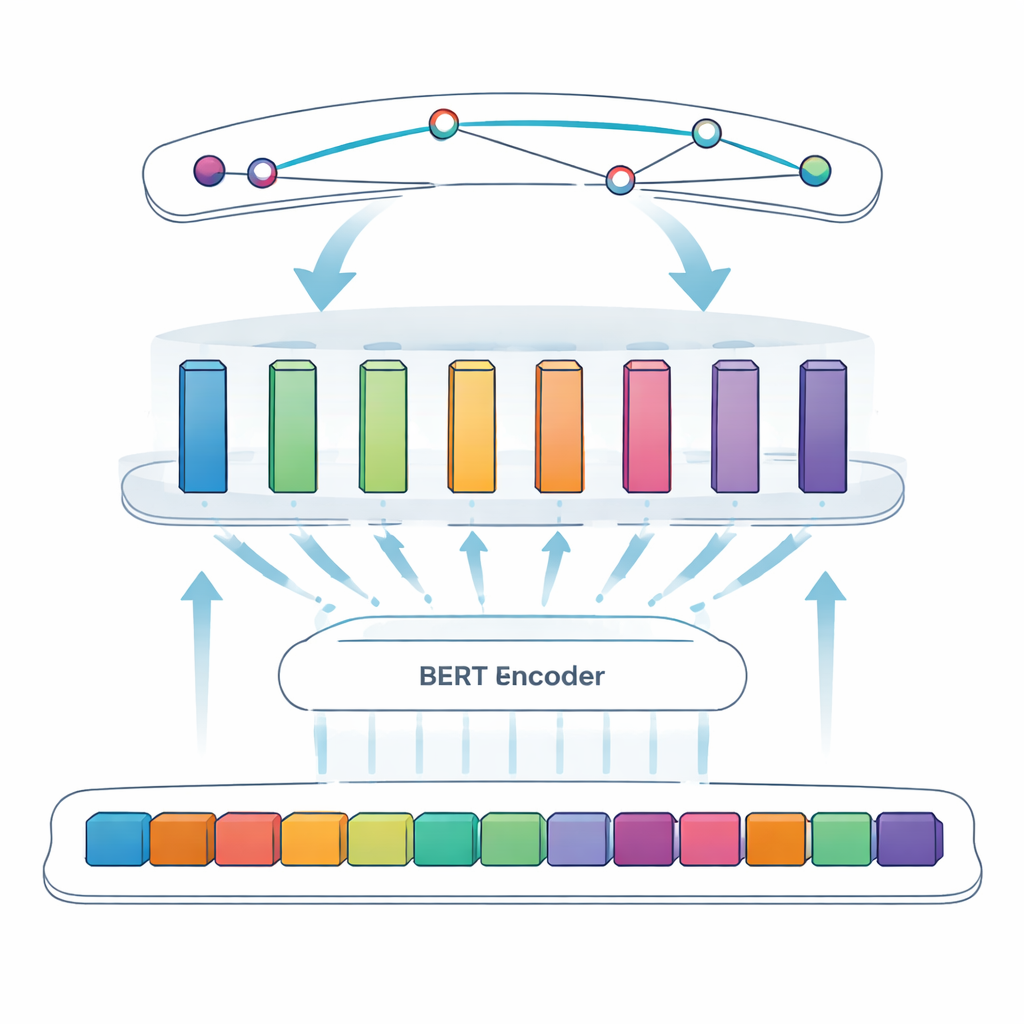
La tarea central de este estudio se denomina reconocimiento de entidades nombradas: conseguir que un ordenador identifique elementos concretos —en este caso, proteínas virales— dentro de texto libre. En enfoques anteriores se confiaba en reglas manuales o listas de términos conocidos, que fallan cuando la nomenclatura es irregular o cambia. Los sistemas más nuevos usan aprendizaje profundo, donde un modelo aprende patrones directamente a partir de ejemplos. El equipo parte de BERT, un modelo de lenguaje que lee el texto en ambas direcciones y aprende representaciones ricas de las palabras según su contexto. Sobre BERT usan un enfoque “SPAN”: en lugar de decidir palabra por palabra si cada token forma parte de una entidad, el modelo predice directamente las posiciones de inicio y fin de cada mención de proteína. Esto facilita manejar casos complejos como nombres anidados o de varias palabras.
Añadir bloques inteligentes al modelo
Para afinar el sistema, los autores introducen una capa feedforward modular adaptativa, que puede verse como varios pequeños módulos expertos intercalados entre BERT y el detector final de spans. Diferentes módulos se especializan en distintos tipos de patrones —por ejemplo, características superficiales como guiones y mezclas de letras y números, o indicios contextuales sutiles que señalan cuándo una letra corta como “S” es en realidad el nombre de una proteína. Un mecanismo de ponderación adaptativa decide entonces, para cada oración, en qué módulos confiar más, combinando sus salidas en una señal más fuerte antes del paso de predicción de spans. Este diseño permite al modelo centrarse de forma flexible en las señales más informativas para cada fragmento de texto.
Poner el enfoque a prueba
Los investigadores compararon rigurosamente su modelo BERT–SPAN modular adaptativo con varios sistemas consolidados, incluido un popular modelo BiLSTM–CRF y una versión estándar de BERT–SPAN sin mejoras modulares. Utilizando medidas estándar de precisión, encontraron que el modelo tradicional BiLSTM–CRF alcanzó una puntuación F1 (el equilibrio entre precisión y recall) de aproximadamente 80,8 % en su conjunto de datos de proteínas de SARS-CoV-2. Cambiar a BERT–SPAN por sí solo elevó el rendimiento al 91,85 %. Añadir la capa modular adaptativa impulsó la F1 hasta 93,66 %, una ganancia relevante en un campo tan maduro. Experimentos de “ablación” cuidadosos —eliminando piezas de la arquitectura una a una— mostraron que tanto la modularización como la ponderación adaptativa contribuyeron a la mejora, especialmente reduciendo errores de delimitación y la confusión entre abreviaturas similares.
Cómo ayuda esto a la investigación futura
Para el lector no especialista, el resultado clave es que los autores construyeron tanto un corpus de alta calidad como un motor de lectura más inteligente capaz de reconocer con fiabilidad las proteínas de SARS-CoV-2 en texto científico. Al convertir un lenguaje desordenado en listas estructuradas de entidades, su sistema facilita mucho la construcción de grafos de conocimiento, el mapeo de qué proteínas virales interactúan con qué moléculas del huésped y el seguimiento de cómo se acumula la evidencia sobre objetivos concretos. En términos prácticos, esto significa que los ordenadores pueden asumir más del trabajo tedioso de minería de texto, liberando a los expertos humanos para interpretar resultados y diseñar experimentos. El estudio muestra que combinar modelos de lenguaje potentes con componentes modulares y adaptativos es una receta prometedora para filtrar la avalancha de literatura biomédica —no solo para la investigación sobre coronavirus, sino para muchos desafíos futuros donde la ciencia supera nuestra capacidad de lectura.
Cita: Shi, X., Fei, Z., Nkhata, A. et al. Optimization and comparison research on adaptive modularization BERT-SPAN model for named entity recognition of SARS-CoV-2 virus proteins. Sci Rep 16, 14207 (2026). https://doi.org/10.1038/s41598-026-44317-7
Palabras clave: proteínas de SARS-CoV-2, reconocimiento de entidades nombradas, minería de texto biomédico, modelo de lenguaje BERT, grafos de conocimiento