Clear Sky Science · it
Ricerca di ottimizzazione e confronto sul modello adattivo modularizzato BERT-SPAN per il riconoscimento delle entità nominate delle proteine del SARS-CoV-2
Perché trovare i nomi delle proteine negli articoli è importante
Ogni settimana compaiono migliaia di nuovi articoli scientifici su COVID-19 e sul suo virus, SARS-CoV-2. Nascosti in questi testi densi ci sono indizi cruciali sulle proteine del virus—macchine molecolari che ne facilitano l’ingresso nelle nostre cellule e che rappresentano bersagli principali per farmaci e vaccini. Ma nessun essere umano può leggere tutto. Questo studio affronta una domanda semplice ma potente: possiamo insegnare ai computer a individuare automaticamente e ad elencare le proteine virali nel testo, con rapidità e precisione sufficienti ad aiutare i ricercatori a restare aggiornati?
Trasformare testi affollati in dati utilizzabili
Quando gli scienziati scrivono di SARS-CoV-2, usano un miscuglio confuso di nomi estesi, abbreviazioni e soprannomi per le sue proteine. La stessa proteina può comparire come “spike protein”, “S” o essere inglobata in frasi come “dominio di legame al recettore dello spike”. Per interpretare questo linguaggio su larga scala, i computer hanno prima bisogno di un «corpus» — una raccolta di frasi accuratamente etichettata in cui gli esperti hanno evidenziato ogni menzione di proteina. Gli autori hanno costruito questa risorsa esaminando circa 200.000 abstract sui coronavirus tratti da PubMed e poi annotando manualmente ogni menzione di proteina SARS-CoV-2 nelle frasi rilevanti. Dopo pulizia e controlli incrociati, hanno ottenuto 7.159 frasi e 12.660 menzioni di proteine etichettate che servono come verità di riferimento per l’addestramento e il test degli algoritmi.
Insegnare alle macchine a leggere i nomi delle proteine virali
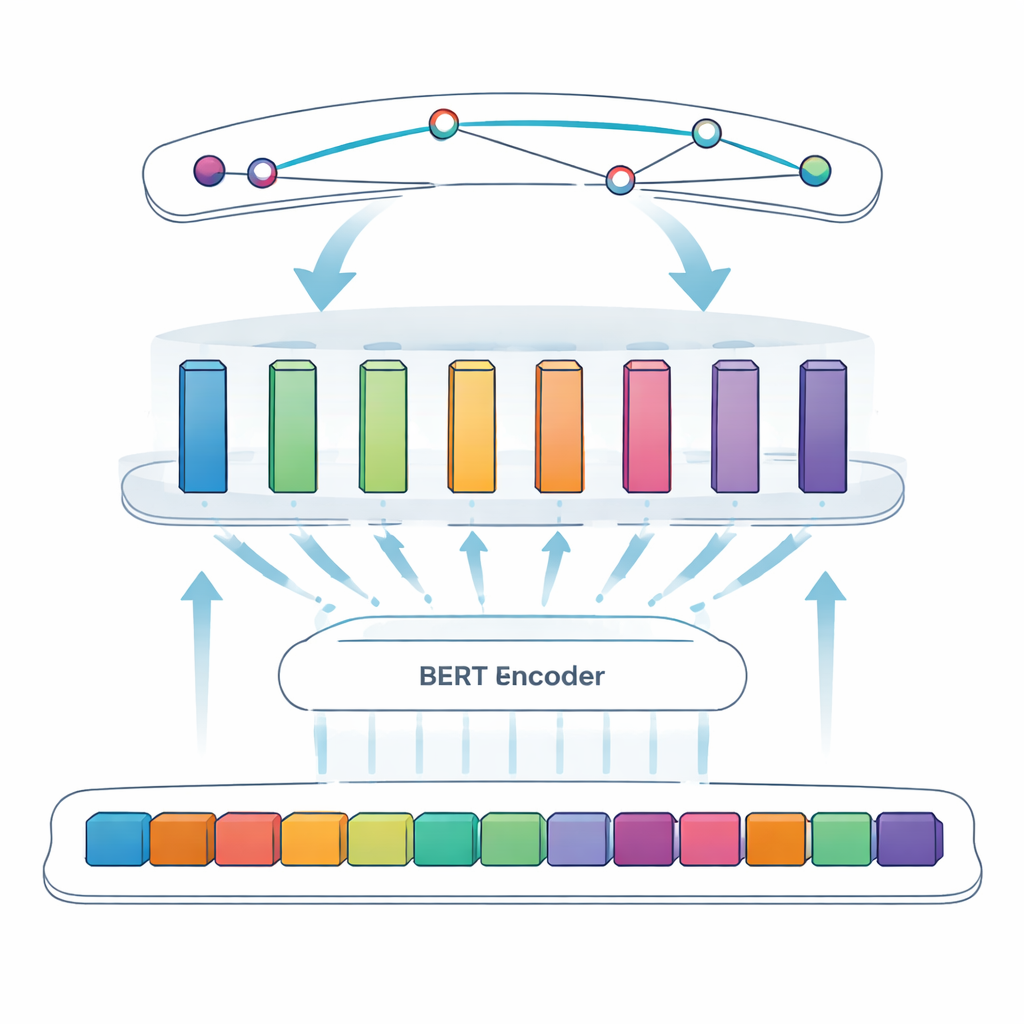
Il compito centrale di questo studio si chiama riconoscimento delle entità nominate: far sì che un computer individui elementi specifici—in questo caso, proteine virali—nel testo libero. Gli approcci precedenti si basavano su regole scritte a mano o su elenchi di termini noti, soluzioni che falliscono quando la nomenclatura è irregolare o evolve. I sistemi più recenti usano il deep learning, in cui un modello apprende i pattern direttamente dagli esempi. Il gruppo si basa su BERT, un modello linguistico che legge il testo in entrambe le direzioni e apprende rappresentazioni ricche delle parole in base al contesto. Sopra BERT usano un approccio “SPAN”: invece di decidere token per token se ciascuno rientra in un’entità, il modello predice direttamente le posizioni di inizio e fine di ogni menzione proteica. Questo facilita la gestione di casi complessi come nomi annidati o costituiti da più parole.
Aggiungere mattoni intelligenti al modello
Per affinare ulteriormente il sistema, gli autori introducono uno strato feedforward modulare adattivo, che può essere visto come diversi piccoli moduli esperti posti tra BERT e il rilevatore finale degli span. Diversi moduli si specializzano in tipi diversi di pattern—per esempio caratteristiche superficiali come trattini e combinazioni di lettere e numeri, o segnali contestuali sottili che indicano quando una lettera breve come “S” è effettivamente il nome di una proteina. Un meccanismo di ponderazione adattiva decide poi, per ogni frase, quali moduli privilegiare, combinando le loro uscite in un segnale complessivo più forte prima della fase di previsione degli span. Questo design permette al modello di concentrarsi in modo flessibile sui segnali più informativi per ciascun pezzo di testo.
Mettere l’approccio alla prova
I ricercatori hanno confrontato rigorosamente il loro modello BERT–SPAN modulare adattivo con diversi sistemi affermati, incluso un diffuso modello BiLSTM–CRF e una versione standard BERT–SPAN senza miglioramenti modulari. Usando misure standard di accuratezza, hanno riscontrato che il tradizionale modello BiLSTM–CRF ha raggiunto un punteggio F1 (bilanciamento tra precisione e richiamo) di circa 80,8 percento sul loro dataset di proteine SARS-CoV-2. Passare a BERT–SPAN ha innalzato la performance al 91,85 percento. L’aggiunta dello strato modulare adattivo ha spinto ulteriormente l’F1 al 93,66 percento, un guadagno significativo in un campo ormai maturo. Esperimenti di «ablation»—rimuovendo pezzi dell’architettura uno alla volta—hanno mostrato che sia la modularizzazione sia la ponderazione adattiva hanno contribuito al miglioramento, specialmente riducendo errori di confine e confusione tra abbreviazioni simili.
Come questo aiuta la ricerca futura
Per un lettore non specialista, il risultato chiave è che gli autori hanno costruito sia un dataset di alta qualità sia un motore di lettura più intelligente in grado di riconoscere in modo affidabile le proteine SARS-CoV-2 nel testo scientifico. Trasformando un linguaggio disordinato in elenchi strutturati di entità, il loro sistema facilita la costruzione di grafi della conoscenza, la mappatura delle interazioni tra proteine virali e molecole dell’ospite e il monitoraggio di come si accumulano le evidenze su specifici bersagli. In termini pratici, ciò significa che i computer possono farsi carico di gran parte del lavoro ripetitivo di text mining, liberando gli esperti umani per concentrarsi sull’interpretazione dei risultati e sulla progettazione di esperimenti. Lo studio mostra che abbinare potenti modelli linguistici a componenti modulari e adattivi è una ricetta promettente per setacciare la massa di letteratura biomedica—non solo per la ricerca sui coronavirus, ma per molte sfide future in cui la scienza supera la nostra capacità di lettura.
Citazione: Shi, X., Fei, Z., Nkhata, A. et al. Optimization and comparison research on adaptive modularization BERT-SPAN model for named entity recognition of SARS-CoV-2 virus proteins. Sci Rep 16, 14207 (2026). https://doi.org/10.1038/s41598-026-44317-7
Parole chiave: proteine SARS-CoV-2, riconoscimento delle entità nominate, estrazione di testi biomedici, modello linguistico BERT, grafi della conoscenza