Clear Sky Science · pl
Projektowanie, synteza i ocena antybakteryjna nowej serii związków pochodnych cyprofloksacyny jako potencjalnych podwójnych inhibitorów gyrazy DNA/topoizomerazy IV
Dlaczego to ma znaczenie w codziennych zakażeniach
Oporność na antybiotyki sprawia, że kiedyś rutynowe zakażenia są trudniejsze i droższe w leczeniu. Cyprofloksacyna, podstawowy antybiotyk stosowany w zakażeniach układu moczowego, jelit i płuc, traci skuteczność wobec niektórych bakterii. Badanie to analizuje sprytny sposób zmodyfikowania konstrukcji cyprofloksacyny, tak by oddziaływała silniej na dwa kluczowe cele u bakterii jednocześnie, co może spowolnić rozwój oporności i poprawić leczenie uporczywych infekcji.
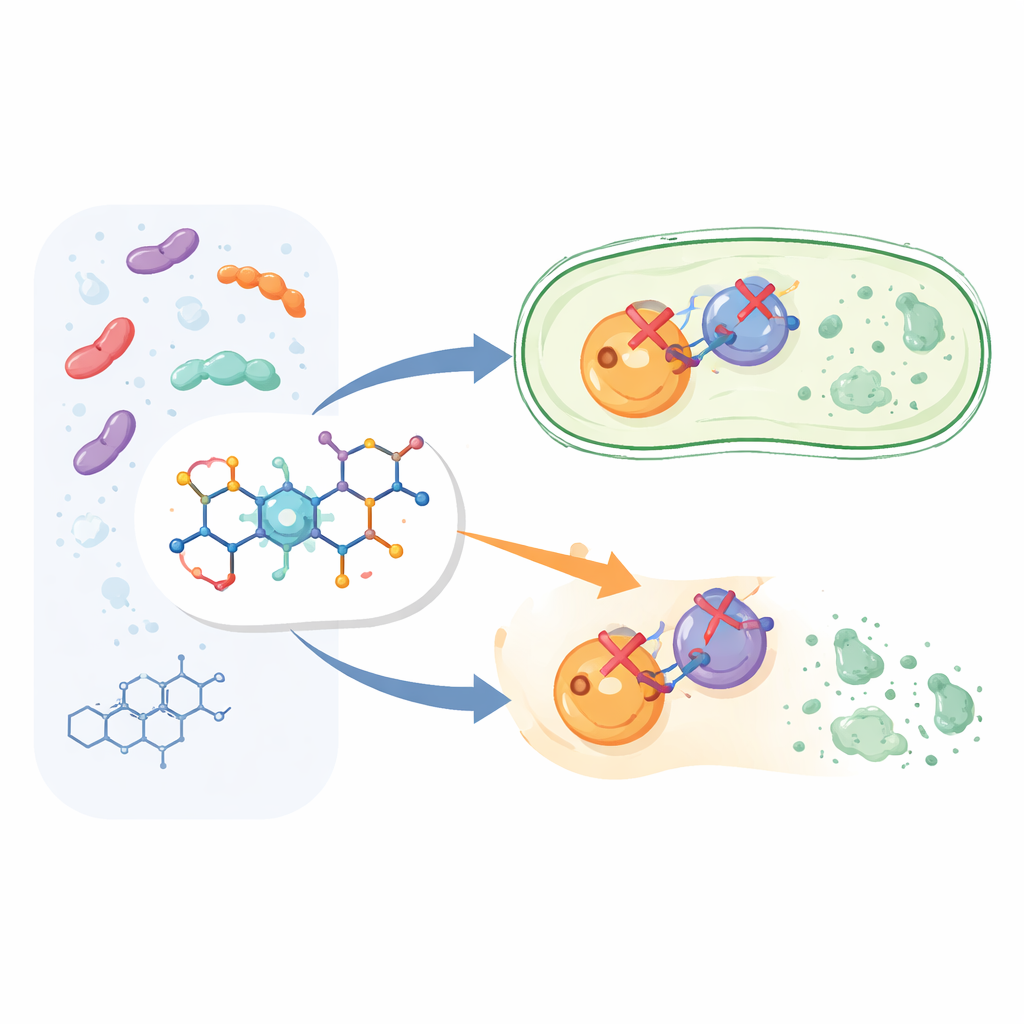
Budowanie mądrzejszej wersji znanego leku
Zamiast zaczynać od całkowicie nowego leku, badacze zmodyfikowali strukturę cyprofloksacyny, przedstawiciela szeroko stosowanej rodziny fluorochinolonów. Zachowali rdzeń cyprofloksacyny, znany z działania bakteriobójczego, a następnie przyłączyli dodatkowy fragment chemiczny zwany arylopirydonem w określonym miejscu cząsteczki. Powstało dwanaście nowych związków oznaczonych jako 6a do 6l. Pomysł polegał na tym, że dodatkowy „uchwyt” pomoże lekowi mocniej i bardziej wszechstronnie wiązać się z bakteryjnymi celami niż standardowa cyprofloksacyna.
Celowanie jednocześnie w dwa bakteryjne punkty podtrzymujące życie
Bakterie polegają na dwóch blisko spokrewnionych enzymach — gyrasie DNA i topoizomerazie IV — aby zarządzać swoim DNA podczas wzrostu i podziału. Cyprofloksacyna już zakłóca działanie tych enzymów, ale nie równomiernie. Nowe związki zaprojektowano tak, by jednocześnie silnie blokować oba cele, co powinno utrudnić bakteriom ucieczkę poprzez mutację tylko jednego enzymu. W testach enzymatycznych z użyciem białek Escherichia coli większość nowych cząsteczek hamowała gyrazę DNA i topoizomerazę IV na użytecznych poziomach. Jeden związek, oznaczony jako 6g, wyróżnił się: nieco silniej blokował gyrazę DNA niż cyprofloksacyna i hamował topoizomerazę IV około siedmiokrotnie skuteczniej, co czyni go najsilniejszą cząsteczką o „podwójnym działaniu” w tym zestawie.
Sprawdzanie nowych cząsteczek przeciwko bakteriom
Zespół następnie sprawdził, czy poprawa aktywności enzymatycznej przekłada się na lepsze działanie przeciwko rzeczywistym bakteriom hodowanym w laboratorium. Wybrane najlepsze związki, w tym 6d, 6f, 6g, 6i i 6l, testowano przeciwko dwóm gatunkom Gram-ujemnym (E. coli i Pseudomonas aeruginosa) oraz dwóm Gram-dodatnim (Staphylococcus aureus i Bacillus subtilis). Ogólnie nowe związki działały najlepiej przeciwko bakteriom Gram-ujemnym, gdzie zdolność do przejścia przez zewnętrzną błonę jest szczególnie istotna. Związek 6g ponownie okazał się faworytem: jego minimalne dawki skuteczne wobec E. coli i P. aeruginosa były w podobnym zakresie co cyprofloksacyny, a także zachowywał umiarkowaną aktywność wobec S. aureus, choć tam był mniej potężny niż lek oryginalny.
Walcząc ze społecznościami bakteryjnymi i badając bezpieczeństwo
Ponad komórkami swobodnie unoszącymi się, bakterie często ukrywają się w śluzowych społecznościach zwanych biofilmami, które sprawiają, że zakażenia są bardziej przewlekłe i trudniejsze do eradikacji. Badacze stwierdzili, że 6g silnie tłumi biofilmy E. coli, redukując je o 96 procent przy tej samej koncentracji potrzebnej do zatrzymania wzrostu, i nadal wykazując znaczną aktywność przy niższych dawkach. Wstępne testy bezpieczeństwa w linii komórek ludzkich gruczołu piersiowego sugerują, że 6g nie jest toksyczny w stężeniach znacznie wyższych niż te potrzebne do działania przeciwbakteryjnego, co jest obiecującym znakiem dla dalszego rozwoju, choć daleko to od pełnej oceny bezpieczeństwa.
Zaglądanie w molekularne uściski dłoni
Aby zrozumieć, dlaczego 6g sprawuje się tak dobrze, zespół użył modelowania komputerowego, by zbadać, jak cząsteczka mieści się w kieszeniach gyrazy DNA i topoizomerazy IV. Symulacje wykazały, że 6g tworzy gęstą sieć oddziaływań — wiązań wodorowych, kontaktów z naładowanymi aminokwasami i ciasnego dopasowania do powierzchni białka — które są silniejsze i bardziej trwałe niż w przypadku cyprofloksacyny. Dodatkowe obliczenia ruchu kompleksów białko–lek w czasie sugerowały, że 6g pomaga utrzymać struktury enzymów w stabilnym, „zablokowanym” stanie, zgodnym z jego silnym hamującym działaniem enzymatycznym. Inne analizy wchłaniania i metabolizmu wskazały na akceptowalny stosunek lipofilności do hydrofilności oraz ograniczoną interakcję z enzymami wątrobowymi, ale też zasugerowały, że związek może nie być idealny do prostego podawania doustnego bez dalszej optymalizacji.
Co to może znaczyć dla przyszłych antybiotyków
Podsumowując, wyniki wyróżniają związek 6g jako obiecujący punkt wyjścia dla antybiotyków nowej generacji. Został zaprojektowany tak, by jednocześnie wyłączać dwa niezbędne enzymy bakteryjne, wykazuje silną aktywność — zwłaszcza przeciwko opornym gatunkom Gram-ujemnym — i zakłóca ochronne biofilmy, przy jednoczesnym braku toksyczności w wstępnym teście na komórkach ludzkich. Jednocześnie jego duże rozmiary i umiarkowane przewidywane wchłanianie sprawiają, że 6g należy traktować raczej jako wiodący szkielet niż gotowy lek. Przy dalszym dopracowaniu pod kątem lepszego wnikania do organizmu i bakterii, podejście o podwójnym działaniu może pomóc wydłużyć użyteczność leków podobnych do cyprofloksacyny i zaoferować nowe opcje w walce z opornymi infekcjami.
Cytowanie: Al-Wahaibi, L.H., Alzahrani, H.A., Bräse, S. et al. Design, synthesis, and antibacterial assessment of a new series of ciprofloxacin-based compounds as possible dual DNA gyrase/topoisomerase IV inhibitors. Sci Rep 16, 13911 (2026). https://doi.org/10.1038/s41598-026-50106-z
Słowa kluczowe: oporność na antybiotyki, pochodne cyprofloksacyny, inhibitory gyrazy DNA, topoizomeraza IV, bakterie Gram-ujemne