Clear Sky Science · pl
Projektowanie, synteza, dokowanie molekularne i ocena cytotoksyczności nowych pochodnych sulfonamidu na bazie pirymidyny jako silnych środków przeciwnowotworowych: wgląd w SAR i profilowanie biologiczne
Dlaczego to ma znaczenie dla przyszłej opieki nad chorymi na raka
Leki chemioterapeutyczne mogą ratować życie, lecz wiele z nich uszkadza tkanki zdrowe niemal tak samo silnie jak guzy, co prowadzi do dotkliwych skutków ubocznych i ogranicza intensywność leczenia, jaką lekarze mogą zastosować. W tej pracy badano nową rodzinę związków syntetycznych zaprojektowanych tak, aby silniej uderzać w komórki nowotworowe niż w zdrowe, z myślą o terapiach skutecznych, a jednocześnie łagodniejszych dla organizmu. Łącząc cechy dwóch dobrze znanych „cegiełek” medycznych, autorzy stworzyli i przetestowali związki, które mogą posłużyć jako punkty wyjścia do opracowania bezpieczniejszych leków przeciwnowotworowych.
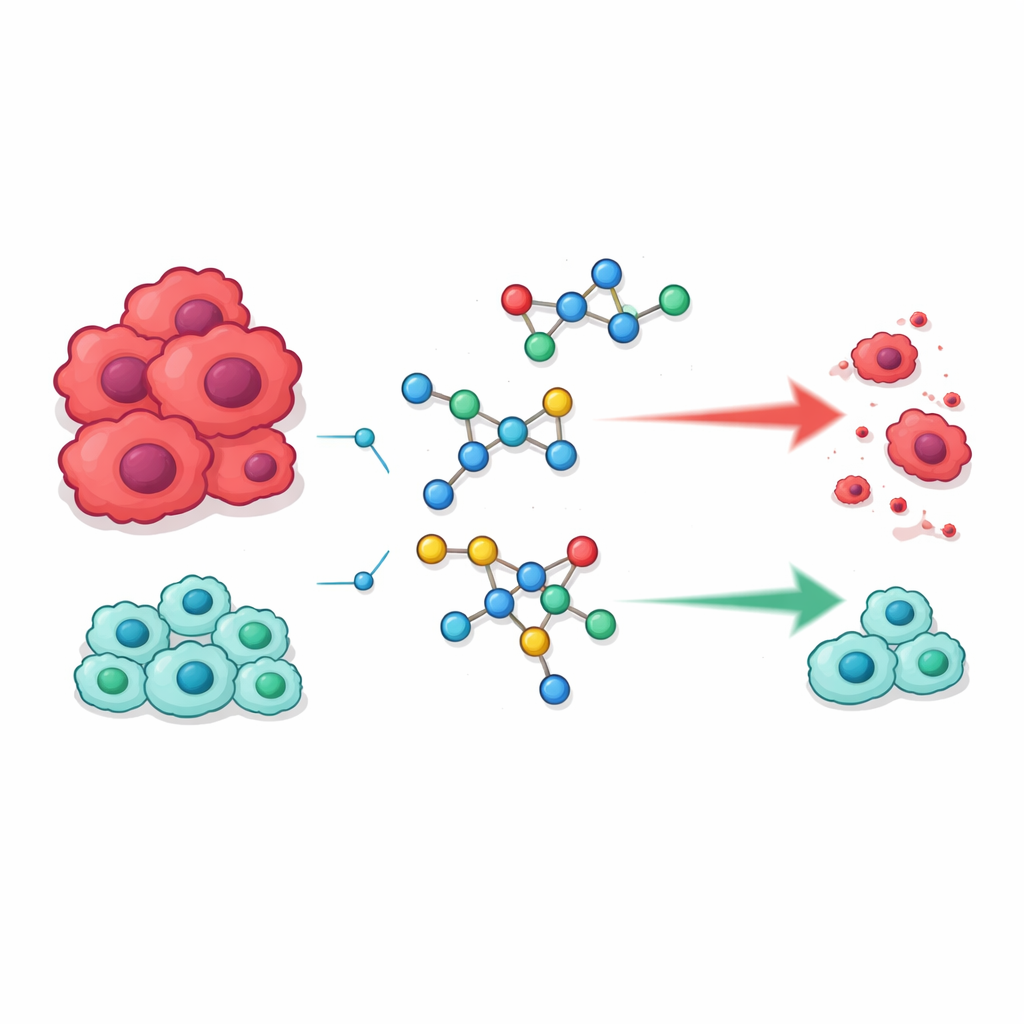
Budowanie nowych cząsteczek z dobrze znanych elementów
Zespół skupił się na dwóch rodzinach chemicznych, które już odgrywają znaczące role we współczesnej medycynie. Jedną z nich są sulfonamidy — wczesna klasa leków pierwotnie znana jako antybiotyki, dziś stosowana także w lekach na schorzenia od jaskry po nadciśnienie i raka. Drugą są pirymidyny — struktury pierścieniowe będące częścią naszego DNA i powszechnie wykorzystywane w lekach przeciwnowotworowych, ponieważ mogą zaburzać kopiowanie materiału genetycznego przez komórki. Pomysł badaczy polegał na połączeniu tych dwóch motywów w pojedyncze hybrydowe cząsteczki, które mogłyby lepiej rozpoznawać i zakłócać procesy niezbędne nowotworom do podziału.
Od syntezy przy ławce laboratoryjnej do biblioteki kandydatów
Stosując etapową syntezę organiczną, naukowcy zbudowali zróżnicowany zestaw sulfonamidów opartych na szkielecie pirymidynowym. Łącząc rdzeń z różnymi układami pierścieni — takimi jak triazole, pyrazole, tiazole i triazyny — dostrajali właściwości takie jak rozkład ładunku elektronowego, kształt i lipofilność. Te cechy wpływają na to, jak dobrze cząsteczka przenika przez błony komórkowe, dociera do celu i unikają uwięzienia lub wypłukania. Każdą nową strukturę dokładnie weryfikowano przy użyciu standardowych narzędzi analizy chemicznej, aby upewnić się, co dokładnie zostało otrzymane przed przejściem do testów biologicznych.
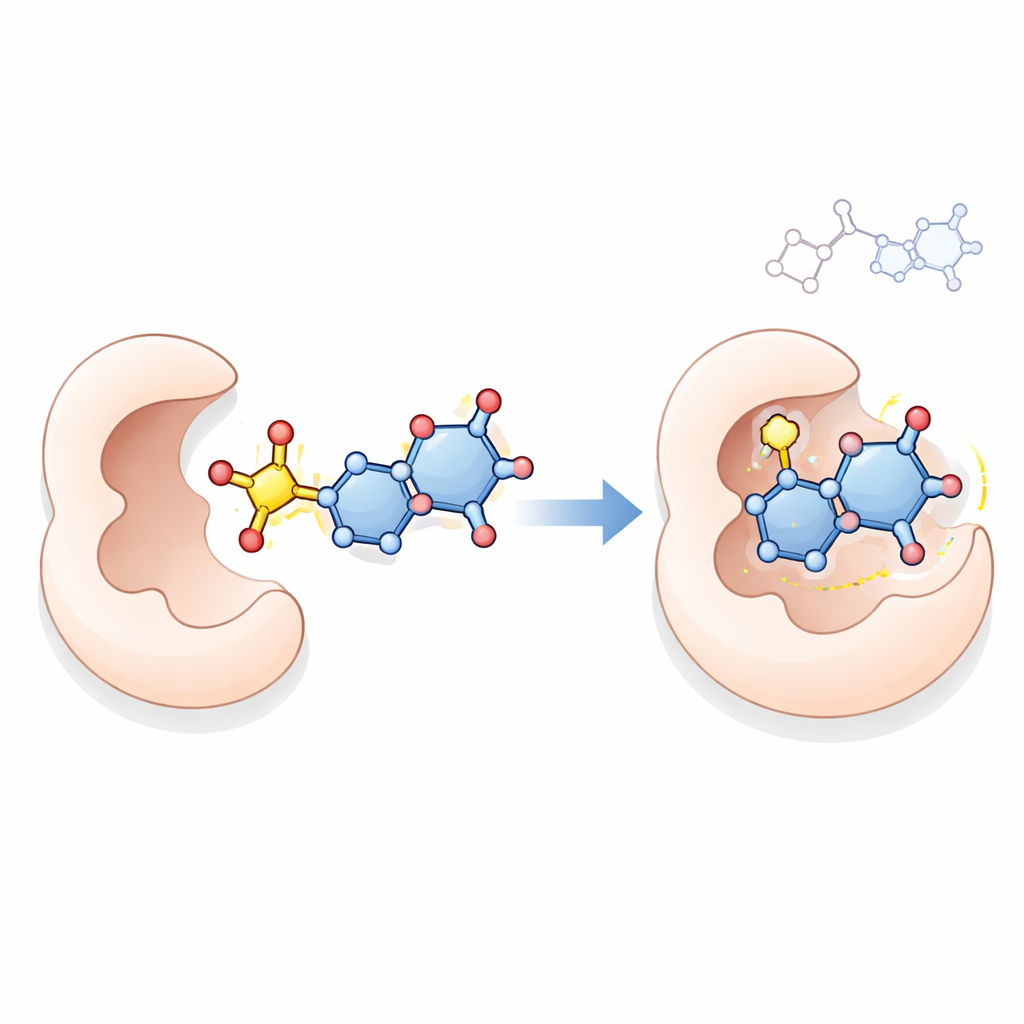
Testowanie nowych związków
Nowe cząsteczki poddano przesiewowi wobec dwóch ludzkich linii komórkowych nowotworowych — HepG2 (wątroba) i MCF‑7 (pierś) — oraz dwóch typów komórek normalnych, fibroblastów płuc WI‑38 i komórek nerkowych VERO, by ocenić zarówno skuteczność, jak i bezpieczeństwo. Kilka kandydatów wyróżniało się spośród reszty. Cztery związki oznaczone jako 18, 21, 23 i 24 zabijały komórki nowotworowe przy stężeniach porównywalnych lub niższych niż w przypadku szeroko stosowanego leku 5‑fluorouracylu. Jednak w przeciwieństwie do 5‑fluorouracylu, te same związki były zauważalnie mniej szkodliwe dla komórek normalnych, co sugeruje korzystniejszą równowagę między skutecznością a toksycznością. Inne człony serii pozbawione pewnych korzystnych cech wykazywały znacznie słabszą aktywność, podkreślając, jak bardzo efekty przeciwnowotworowe zależą od drobnych zmian strukturalnych.
Czym różnią się najsilniejsze kandydaty
Porównując struktury i wyniki testów, autorzy wyprowadzili wstępne zasady dotyczące tego, co działa najlepiej. Najbardziej aktywne związki miały tendencję do noszenia niewielkich grup oddających gęstość elektronową do rdzenia, co poprawiało interakcje cząsteczek z ich biologicznymi celami. Wykazywały też „właściwy” poziom lipofilności — wystarczającą afinność do lipidów, by przenikać przez błony komórkowe, ale nie tak dużą, by niespecyficznie przyczepiać się do wszystkiego. Dodatkowo ich kształty unikały dużych przeszkód w rejonach aktywnych, pozwalając na dopasowanie w kieszeniach wiązania. Te cechy wydawały się zwiększać zarówno cytotoksyczność wobec komórek nowotworowych, jak i oszczędzanie komórek normalnych, sugerując, że cząsteczki mogą celować bardziej selektywnie w ścieżki, od których zależą guzy.
Bliższe spojrzenie na możliwy mechanizm działania
Aby zbadać możliwy tryb działania, zespół zastosował komputerowe dokowanie molekularne — technikę symulującą, jak mała cząsteczka może ulokować się w miejscu aktywnym białka. Zbadano interakcje czołowych związków 18, 21, 23 i 24 z syntazą tymidylanową, kluczowym enzymem potrzebnym komórkom nowotworowym do syntezy DNA i znanym celem leków takich jak 5‑fluorouracyl. Symulacje sugerowały, że te nowe cząsteczki, zwłaszcza związek 23, mogą tworzyć wielokrotne stabilizujące kontakty w obrębie kieszeni enzymu, w niektórych przypadkach wiążąc się silniej niż lek referencyjny. Choć samo dokowanie nie dowodzi działania w żywych komórkach, daje wiarygodne wyjaśnienie zgodne z zaobserwowanym silnym efektem antyproliferacyjnym.
Co to oznacza na przyszłość
Podsumowując, praca przedstawia cztery molekuły prowadzące, które łączą silną aktywność wobec komórek raka wątroby i piersi z łagodniejszym wpływem na komórki normalne, wraz z wczesnymi wskazówkami na temat cech strukturalnych determinujących tę równowagę. Mówiąc prościej, badacze naszkicowali chemiczny plan działania dla kandydatów do chemioterapii następnej generacji, które mogą być skuteczne i mniej szkodliwe dla tkanek zdrowych. Konieczne będą dalsze badania na poziomie enzymów, całych komórek, a ostatecznie na modelach zwierzęcych, lecz te hybrydy pirymidyno‑sulfonamidowe stanowią obiecujący krok w kierunku bardziej ukierunkowanych i lepiej tolerowanych terapii przeciwnowotworowych.
Cytowanie: Bayoumy, N.M., Fadda, A.A., Gaffer, H.E. et al. Design, synthesis, molecular docking and cytotoxic evaluation of novel pyrimidine-based sulfonamide derivatives as potent anticancer agents: SAR insights and biological profiling. Sci Rep 16, 9820 (2026). https://doi.org/10.1038/s41598-026-41711-z
Słowa kluczowe: leki przeciwnowotworowe, sulfonamidy, hybrydy pirymidynowe, syntaza tymidylanowa, dokowanie molekularne