Clear Sky Science · en
Design, synthesis, molecular docking and cytotoxic evaluation of novel pyrimidine-based sulfonamide derivatives as potent anticancer agents: SAR insights and biological profiling
Why this matters for future cancer care
Chemotherapy drugs can be lifesaving, but many damage healthy tissues nearly as much as tumors, leading to harsh side effects and limiting how aggressively doctors can treat cancer. This study explores a new family of lab‑made molecules designed to hit cancer cells harder than healthy cells, aiming for treatments that are both powerful and gentler on the body. By blending features from two well‑known medicine “building blocks,” the researchers created and tested compounds that may serve as starting points for safer anticancer drugs.
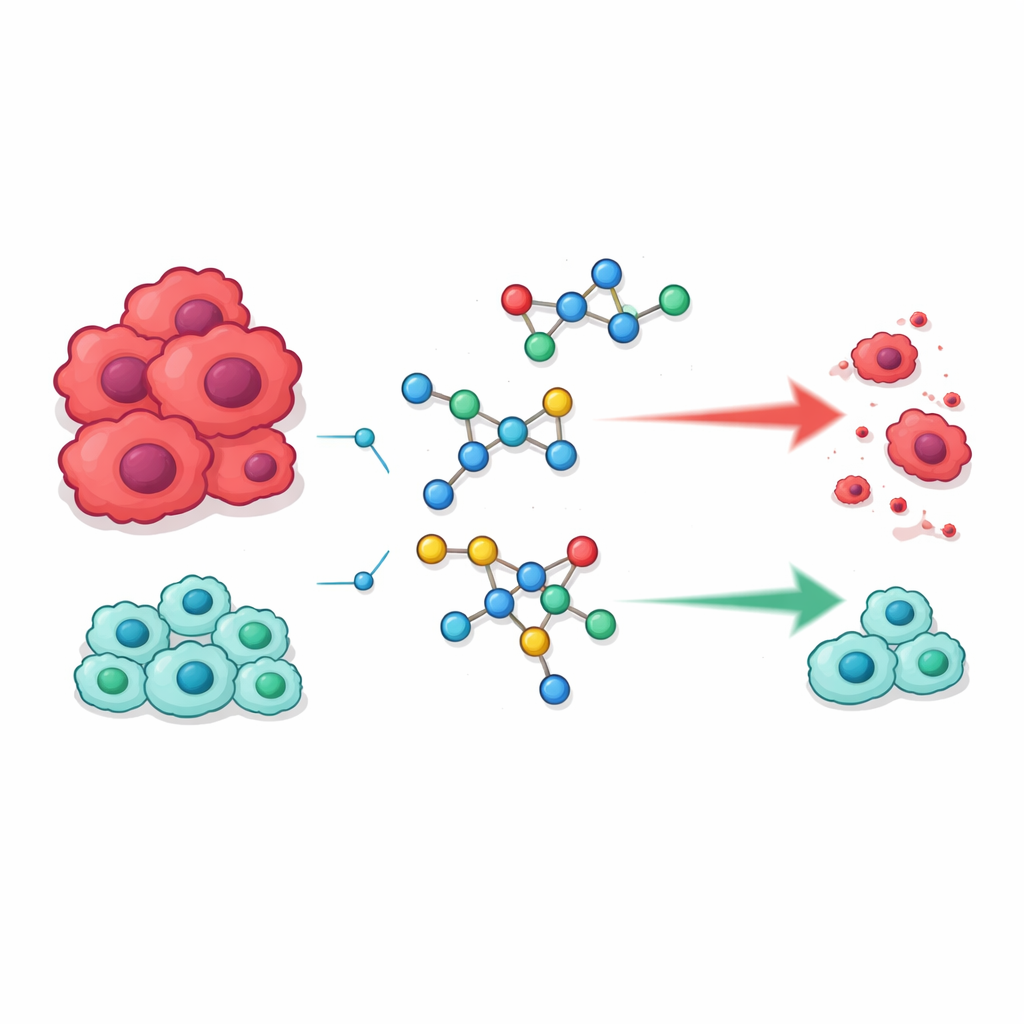
Building new molecules from familiar pieces
The team focused on two chemical families that already play starring roles in modern medicine. One is sulfonamides, an early class of drugs originally famous as antibiotics but now found in medicines for conditions ranging from glaucoma to high blood pressure and cancer. The other is pyrimidines, ring‑shaped structures that form part of our DNA and are widely used in anticancer drugs because they can interfere with how cells copy their genetic material. The researchers’ idea was to fuse these two motifs into single hybrid molecules that might better recognize and disrupt processes cancer cells rely on to divide.
From bench chemistry to a library of candidates
Using stepwise organic synthesis, the scientists built a diverse set of pyrimidine‑based sulfonamide compounds. By linking the core framework to different surrounding ring systems—such as triazoles, pyrazoles, thiazoles, and triazines—they tuned properties like electronic charge, shape, and fat‑solubility. These features help determine how well a molecule can cross cell membranes, reach its target, and avoid becoming trapped or washed out. Each new structure was carefully verified using standard tools of chemical analysis, ensuring the researchers knew exactly what they had made before moving on to biological tests.
Putting the new compounds to the test
The new molecules were screened against two human cancer cell lines—HepG2 (liver) and MCF‑7 (breast)—as well as two types of normal cells, WI‑38 lung fibroblasts and VERO kidney cells, to assess both potency and safety. Several candidates stood out. Four compounds, labeled 18, 21, 23, and 24, killed cancer cells at concentrations similar to or lower than those needed for the widely used drug 5‑fluorouracil. Yet, unlike 5‑fluorouracil, these same compounds were noticeably less harmful to normal cells, suggesting a more favorable balance between efficacy and toxicity. Other members of the series that lacked certain helpful features showed much weaker activity, underscoring how sensitive anticancer effects are to small structural changes.
What makes the strongest candidates different
By comparing structures and test results, the authors pieced together early rules about what works best. The most active compounds tended to carry small groups that donate electron density to the core scaffold, improving how the molecules interact with their biological targets. They also showed a “just right” level of lipophilicity—sufficiently attracted to fats to slip through cell membranes, but not so greasy that they stick nonspecifically to everything. In addition, their shapes avoided bulky obstacles near the active regions, allowing a snug fit into binding sites. These traits appeared to boost both cancer‑cell killing power and sparing of normal cells, hinting that the molecules may be homing in more selectively on pathways tumors depend on.
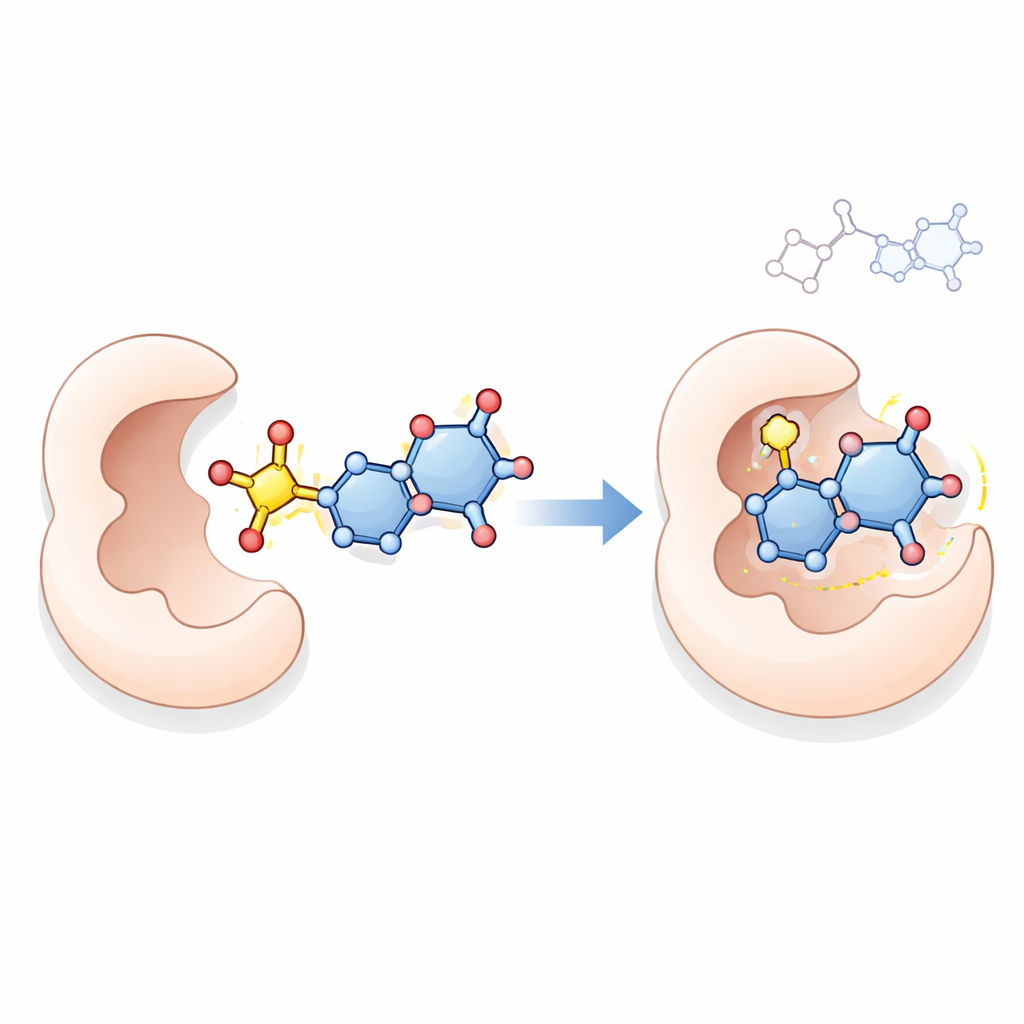
A closer look at how they might work
To explore a possible mode of action, the team used computer‑based molecular docking, a technique that simulates how a small molecule might sit inside a protein’s active site. They examined how top compounds 18, 21, 23, and 24 interact with thymidylate synthase, a key enzyme cancer cells need to make DNA and a known target of drugs like 5‑fluorouracil. The simulations suggested that these new molecules, especially compound 23, can form multiple stabilizing contacts within the enzyme’s pocket, in some cases binding more strongly than the reference drug. While docking alone cannot prove how the compounds work inside living cells, it offers a plausible explanation consistent with their strong antiproliferative effects.
What this means going forward
Overall, this work delivers four lead molecules that combine potent activity against liver and breast cancer cells with milder effects on normal cells, along with early clues about the structural features that drive this balance. In everyday terms, the researchers have sketched a chemical blueprint for next‑generation chemotherapy candidates that may be both effective and kinder to healthy tissue. Further studies in enzymes, whole cells, and eventually animals will be needed, but these pyrimidine‑sulfonamide hybrids mark a promising step toward more targeted and tolerable cancer treatments.
Citation: Bayoumy, N.M., Fadda, A.A., Gaffer, H.E. et al. Design, synthesis, molecular docking and cytotoxic evaluation of novel pyrimidine-based sulfonamide derivatives as potent anticancer agents: SAR insights and biological profiling. Sci Rep 16, 9820 (2026). https://doi.org/10.1038/s41598-026-41711-z
Keywords: anticancer drugs, sulfonamides, pyrimidine hybrids, thymidylate synthase, molecular docking