Clear Sky Science · pl
Wyciszenie PCDHGC3 wspiera przerzuty jasnokomórkowego raka nerki poprzez aktywację mTOR/HIF2α, przestawienie metabolizmu lipidów i unikanie ferroptozy
Dlaczego to badanie nad rakiem nerki ma znaczenie
Jasnokomórkowy rak nerki jest najczęstszą postacią raka nerek i jest znany ze skłonności do przerzutów oraz oporności na leczenie. W pracy tej ujawniono, w jaki sposób utrata pojedynczego białka błonowego o nazwie PCDHGC3 ułatwia wzrost guza, jego rozprzestrzenianie się i unikanie formy śmierci komórkowej związanej z uszkodzeniem lipidów. Śledząc łańcuch zdarzeń od błony komórkowej po zmianę metabolizmu tłuszczów w komórkach nowotworowych, badanie wskazuje nowe terapie skojarzone, które mogłyby uczynić zaawansowane raki nerki bardziej podatnymi na leczenie.
Brakujący „hamulec” komórkowy w guzach nerek
Naukowcy zaczęli od analizy dużych zbiorów danych pacjentów, aby sprawdzić zachowanie rodziny genów odpowiedzialnych za adhezję komórkową w jasnokomórkowym raku nerki. Spośród kilkudziesięciu powiązanych genów PCDHGC3 wyróżniał się jako wyraźnie aktywny w tkance prawidłowej, lecz obniżony w agresywnych guzach. Pacjenci, których guzy miały niskie poziomy tego genu, częściej mieli zaawansowaną chorobę i krótsze przeżycie. W przeciwieństwie do wielu krewniaczych genów, PCDHGC3 nie było jedynie wyciszane przez silne znakowanie chemiczne DNA, co sugeruje odrębny mechanizm regulacji i możliwą unikalną rolę ochronną w komórkach nerkowych.
Od wolno rosnących do inwazyjnych komórek nowotworowych
Aby sprawdzić funkcję PCDHGC3, zespół obniżył jego poziomy w dwóch liniach ludzkich komórek raka nerki i porównał je z komórkami kontrolnymi. Po wyciszeniu tego genu komórki dzieliły się szybciej, przemieszczały się szybciej przez cykl komórkowy i wygrywały rywalizację z komórkami kontrolnymi w tych samych hodowlach. W trójwymiarowych modelach nowotworów drukowanych biotechnologicznie, które lepiej odzwierciedlają tkankę, komórki pozbawione PCDHGC3 tworzyły większe, gęstsze struktury. U myszy guzy bez tego genu rosły ponad czterokrotnie większe i wykazywały więcej aktywnie dzielących się komórek. Zmienione komórki przeszły także od zwartego, nabłonkowego kształtu w stronę bardziej plastycznej, migracyjnej formy związanej z inwazją, i chętniej tworzyły odległe guzy po wstrzyknięciu do krwiobiegu.
Szlak wzrostu włączony na pełne obroty
Analiza sygnalizacji wykazała, że utrata PCDHGC3 aktywuje znany regulator wzrostu w komórce o nazwie mTOR, wraz z powiązanymi białkami napędzającymi syntezę białek i przeżycie. Kluczowa kinaza adhezji ogniskowej na powierzchni komórki także stała się bardziej aktywna, łącząc zmiany na błonie z wewnętrznym przełącznikiem wzrostu. Ważnym następstwem było nieprawidłowe gromadzenie się czynnika HIF2α, który pomaga komórkom nowotworowym adaptować się do niedoboru tlenu, wspiera tworzenie naczyń i zmienia ich metabolizm. Leki blokujące mTOR lub HIF2α spowalniały nadmierny wzrost komórek pozbawionych PCDHGC3, zarówno w hodowlach dwuwymiarowych, jak i w modelach 3D, a także zmniejszały przerzuty w modelu myszy z guzem wszczepionym do nerki, co sugeruje, że ten szlak jest kluczowy dla agresywnego zachowania.
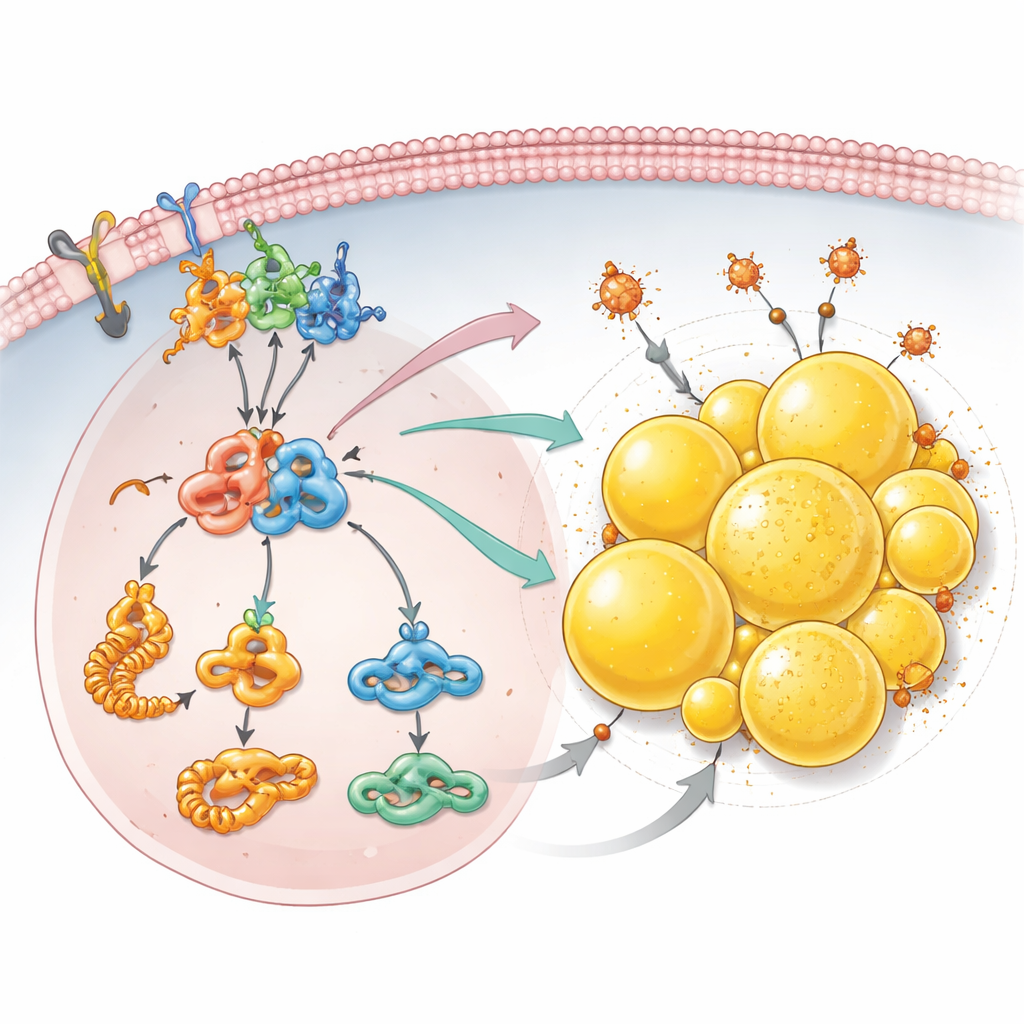
Magazynowanie tłuszczu jako tarcza przeciw śmierci komórki
Ponad sygnałami wzrostu zespół odkrył, że utrata PCDHGC3 przekształca sposób, w jaki komórki nowotworowe gospodarują tłuszczami. Szczegółowe analizy białek wykazały zwiększoną produkcję prekursorów kwasów tłuszczowych oraz wyższe poziomy acetylo-CoA, centralnego substratu wykorzystywanego do syntezy lipidów. Komórki pozbawione PCDHGC3 zgromadziły znacznie więcej kropelek lipidowych — małych, wypełnionych tłuszczem sfer wewnątrzkomórkowych, które są szczególnie liczne w tym typie raka nerki. To nagromadzenie zależało od osi mTOR–HIF2α oraz od PLIN2, białka pokrywającego kropelki, którego poziomy wzrosły po wyciszeniu PCDHGC3. Te kropelki robiły więcej niż tylko magazynować nadmiar tłuszczu: pomagały komórkom uciec przed ferroptozą, formą śmierci komórkowej napędzaną przez żelazo i uszkodzone lipidy. Gdy zmniejszono poziom PLIN2, zwłaszcza w komórkach już pozbawionych PCDHGC3, kropelki lipidowe zmalały, a komórki stały się znacznie bardziej wrażliwe na lek wywołujący ferroptozę.
Przekształcenie triku przetrwania guza w jego słabość
Podsumowując, badanie ujawnia łańcuch zdarzeń, w którym utrata PCDHGC3 na powierzchni komórki aktywuje kinazę adhezji ogniskowej, włącza mTOR i HIF2α, zwiększa syntezę tłuszczów i buduje ochronne kropelki lipidowe pokryte PLIN2. Ta sieć nie tylko napędza wzrost i rozprzestrzenianie się guza, ale także pozwala komórkom nowotworowym ukrywać wrażliwe lipidy przed destrukcyjnymi reakcjami, które w przeciwnym razie by je zabiły. Autorzy proponują, że ukierunkowanie nowo zdefiniowanej osi PCDHGC3–mTOR–HIF2α–PLIN2 może stanowić wielotorową strategię dla zaawansowanego jasnokomórkowego raka nerki: łączenie inhibitorów mTOR lub HIF2α z lekami indukującymi ferroptozę, a potencjalnie hamowanie PLIN2, może obrócić metaboliczną adaptację guza w jego piętę achillesową.
Cytowanie: Celada, L., Cubiella, T., San-Juan-Guardado, J. et al. PCDHGC3 silencing promotes clear cell renal cell carcinoma metastasis via mTOR/HIF2α activation, lipid metabolism rewiring, and ferroptosis evasion. Cell Death Dis 17, 409 (2026). https://doi.org/10.1038/s41419-026-08643-y
Słowa kluczowe: jasnokomórkowy rak nerki, PCDHGC3, szlak mTOR HIF2alpha, kropelki lipidowe, ferroptoza