Clear Sky Science · de
PCDHGC3-Stummschaltung fördert Metastasierung von klarzelligem Nierenzellkarzinom über mTOR/HIF2α-Aktivierung, Umbau des Lipidstoffwechsels und Vermeidung von Ferroptose
Warum diese Nierenkrebsforschung wichtig ist
Das klarzellige Nierenzellkarzinom ist die häufigste Form von Nierenkrebs und dafür bekannt, in andere Organe zu streuen und Therapien zu widerstehen. Diese Studie zeigt auf, wie der Verlust eines einzigen Zelloberflächenproteins, PCDHGC3 genannt, Tumoren im Nieregewebe beim Wachsen, Ausbreiten und beim Umgehen einer Form des Zelltods, die mit Lipidschäden zusammenhängt, unterstützt. Indem die Forscher diese Kaskade vom Zellmembranniveau bis hin zur Fetthandhabung der Krebszellen nachverfolgen, weisen die Ergebnisse auf neue Kombinationsbehandlungen hin, die fortgeschrittene Nierenkrebserkrankungen anfälliger für Therapien machen könnten.
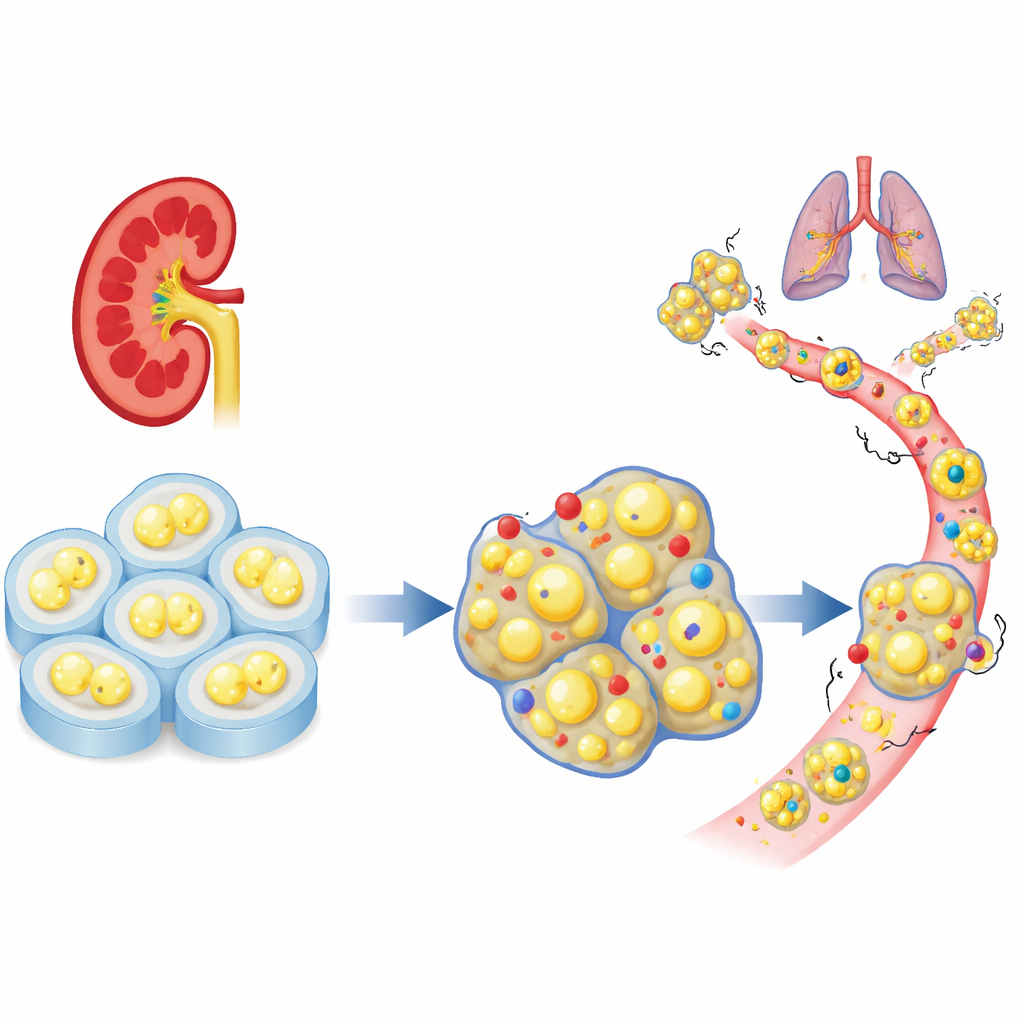
Eine fehlende zelluläre „Bremse“ in Nierentumoren
Die Forschenden begannen mit der Auswertung großer Patientendatensätze, um zu prüfen, wie sich eine Familie von Zelladhäsionsgenen beim klarzelligen Nierenkrebs verhält. Unter Dutzenden verwandter Gene fiel PCDHGC3 auf: Es war in normalem Gewebe ungewöhnlich hoch exprimiert, in aggressiven Tumoren dagegen vermindert. Patienten mit niedrigen Tumorwerten dieses Gens hatten häufiger fortgeschrittene Erkrankungen und eine kürzere Überlebenszeit. Anders als viele Verwandte wurde PCDHGC3 nicht einfach durch starke chemische Markierung der DNA ausgeschaltet, was darauf hindeutet, dass es anders reguliert wird und in Nierenzellen eine besondere schützende Rolle spielen könnte.
Von langsam wachsend zu invasiven Krebszellen
Um die Funktion von PCDHGC3 zu testen, reduzierten die Wissenschaftler das Gen in zwei menschlichen Nierenkrebszelllinien und verglichen diese mit Kontrollzellen. Nach der Stilllegung teilten sich die Zellen schneller, durchliefen den Zellzyklus zügiger und setzten sich gegenüber normalen Gegenstücken in gemeinsamen Kulturen besser durch. In 3D-bioprinteten Tumormodellen, die echtes Gewebe besser nachahmen, bildeten PCDHGC3-defiziente Zellen größere, dichtere Strukturen. In Mäusen wuchsen Tumoren ohne dieses Gen mehr als viermal so groß und zeigten mehr aktiv teilende Zellen. Diese veränderten Zellen wechselten außerdem von einer kompakten, epithelialen Form zu einer flexibleren, mobileren Gestalt, die mit Invasion assoziiert ist, und sie bildeten nach Injektion in den Blutkreislauf leichter Fernmetastasen.
Ein Wachstumsweg, der auf Hochtouren läuft
Bei der Untersuchung der Signalwege fanden die Forschenden heraus, dass der Verlust von PCDHGC3 einen bekannten zellulären Wachstumsregulator namens mTOR sowie verwandte Proteine, die Proteinsynthese und Überleben antreiben, aktiviert. Eine wichtige Adhäsionskinase an der Zelloberfläche, die focal adhesion kinase, wurde ebenfalls stärker aktiv, wodurch Veränderungen an der Membran mit diesem inneren Wachstumsschalter verbunden wurden. Eine besonders relevante Folge war die abnorme Anhäufung eines Faktors namens HIF2α, der Tumorzellen hilft, sich an niedrige Sauerstoffbedingungen anzupassen, die Gefäßbildung zu fördern und ihren Stoffwechsel umzubauen. Medikamente, die mTOR oder HIF2α blockieren, verlangsamten jeweils das Überwachsen von PCDHGC3-defizienten Zellen in zweidimensionalen Kulturen und in 3D-Modellen und verringerten zudem metastatische Tumoren in einem in die Niere eingebetteten Mausmodell, was darauf hindeutet, dass dieser Signalweg zentral für das aggressive Verhalten ist.
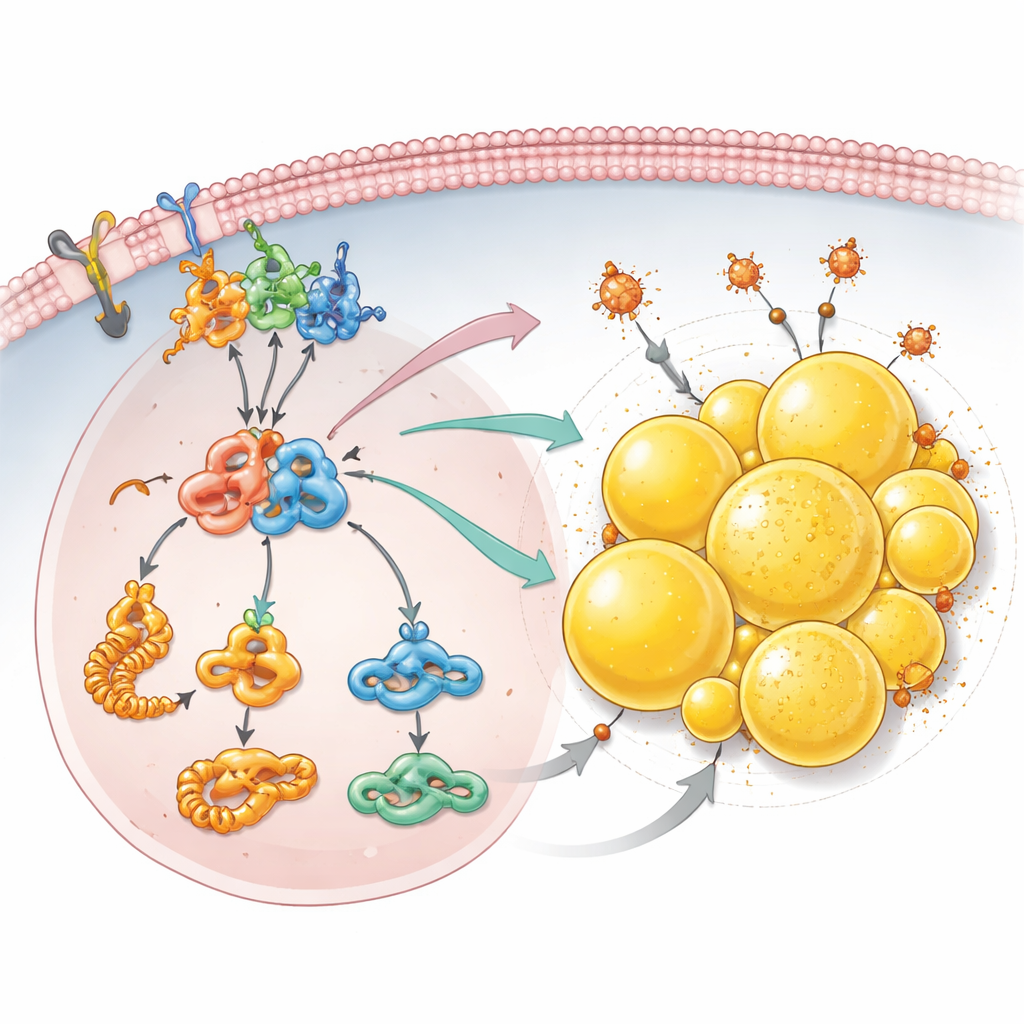
Fettspeicherung als Schutz vor Zelltod
Über die Wachstumssignale hinaus entdeckte das Team, dass der Verlust von PCDHGC3 den Umgang der Krebszellen mit Fetten umprogrammiert. Detaillierte Proteinanalyse zeigte eine erhöhte Produktion von Bausteinen für Fettsäuren und höhere Mengen an Acetyl‑CoA, einem zentralen Brennstoff für die Lipidsynthese. Zellen ohne PCDHGC3 bauten deutlich mehr Lipidtröpfchen auf – kleine, mit Fett gefüllte Kugeln im Zellinneren, die in diesem Nierentumortyp besonders zahlreich sind. Diese Ansammlung hing vom mTOR–HIF2α-Achse und von PLIN2 ab, einem Tröpfchen-ummantelnden Protein, dessen Spiegel nach PCDHGC3-Stummschaltung anstiegen. Diese Tröpfchen dienten nicht nur der Speicherung überschüssiger Fette: Sie halfen den Zellen, der Ferroptose zu entkommen, einer Form des Zelltods, die von Eisen und geschädigten Lipiden getrieben wird. Wurde PLIN2 reduziert, insbesondere in Zellen ohne PCDHGC3, schrumpften die Lipidtröpfchen und die Zellen wurden deutlich empfindlicher gegenüber einem Medikament, das Ferroptose auslöst.
Den Überlebens-Trick des Tumors zur Schwäche machen
Insgesamt zeigt die Studie eine Abfolge von Ereignissen: Der Verlust von PCDHGC3 an der Zelloberfläche aktiviert die focal adhesion kinase, schaltet mTOR und HIF2α ein, steigert die Fettsynthese und erzeugt schützende, von PLIN2 umhüllte Lipidtröpfchen. Dieses Netzwerk treibt nicht nur Tumorwachstum und -ausbreitung, sondern ermöglicht es Krebszellen auch, anfällige Lipide vor zerstörerischen Reaktionen zu verbergen, die sie sonst töten würden. Die Autorinnen und Autoren schlagen vor, dass die neu definierte PCDHGC3–mTOR–HIF2α–PLIN2-Achse einen vielschichtigen Ansatz für fortgeschrittenes klarzelliges Nierenzellkarzinom bieten könnte: Die Kombination von mTOR- oder HIF2α-Inhibitoren mit Ferroptose-auslösenden Wirkstoffen und gegebenenfalls PLIN2-Blockade könnte die metabolische Anpassung des Tumors in eine Achillesferse verwandeln.
Zitation: Celada, L., Cubiella, T., San-Juan-Guardado, J. et al. PCDHGC3 silencing promotes clear cell renal cell carcinoma metastasis via mTOR/HIF2α activation, lipid metabolism rewiring, and ferroptosis evasion. Cell Death Dis 17, 409 (2026). https://doi.org/10.1038/s41419-026-08643-y
Schlüsselwörter: klarzelliges Nierenzellkarzinom, PCDHGC3, mTOR HIF2alpha Signalweg, Lipidtröpfchen, Ferroptose