Clear Sky Science · en
PCDHGC3 silencing promotes clear cell renal cell carcinoma metastasis via mTOR/HIF2α activation, lipid metabolism rewiring, and ferroptosis evasion
Why this kidney cancer research matters
Clear cell renal cell carcinoma is the most common form of kidney cancer and is notorious for spreading to other organs and resisting treatment. This study uncovers how the loss of a single cell-surface protein, called PCDHGC3, helps kidney tumors grow, spread, and dodge a form of cell death linked to fat damage. By tracing this chain of events from the cell membrane down to how cancer cells handle fats, the work points to new combination therapies that could make advanced kidney cancers more vulnerable to treatment.
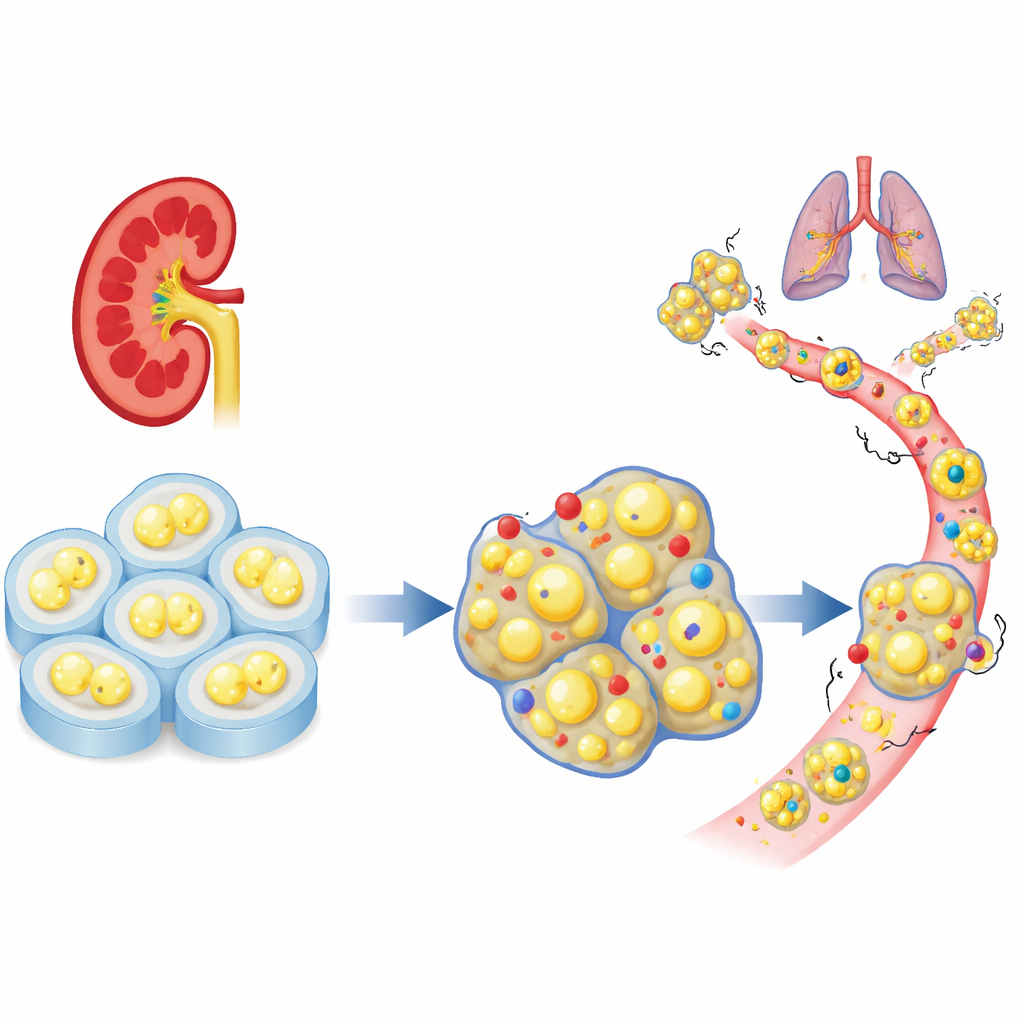
A missing cell “brake” in kidney tumors
The researchers began by looking at large patient datasets to see how a family of cell-adhesion genes behaves in clear cell kidney cancer. Among dozens of related genes, PCDHGC3 stood out as unusually active in normal tissue but reduced in aggressive tumors. Patients whose tumors had low levels of this gene were more likely to have advanced disease and shorter survival. Unlike many of its relatives, PCDHGC3 did not simply get shut off by heavy chemical tagging of its DNA, suggesting it is regulated in a distinct way and may play a unique protective role in kidney cells.
From slow-growing to invasive cancer cells
To test what PCDHGC3 actually does, the team reduced its levels in two human kidney cancer cell lines and compared them with control cells. When this gene was silenced, cells divided faster, moved through the cell cycle more quickly, and outcompeted normal counterparts grown in the same dish. In 3D bioprinted tumor models that better mimic real tissue, PCDHGC3-deficient cells formed larger, denser structures. In mice, tumors lacking this gene grew over four times bigger and showed more actively dividing cells. These altered cells also shifted from a compact, epithelial shape toward a more flexible, migratory form associated with invasion, and they more readily formed distant tumors after being injected into the bloodstream.
A growth pathway switched into overdrive
Diving into the signaling machinery, the researchers found that the loss of PCDHGC3 activates a well-known growth controller inside cells called mTOR, along with related proteins that drive protein synthesis and survival. A key adhesion enzyme at the cell surface, focal adhesion kinase, also became more active, linking changes at the membrane to this internal growth switch. One particularly important consequence was the abnormal buildup of a factor called HIF2α, which helps tumor cells adapt to low oxygen, support blood vessel growth, and change their metabolism. Drugs that block mTOR or HIF2α each slowed the overgrowth of PCDHGC3-deficient cells, both in flat cultures and in 3D models, and also reduced metastatic tumors in a kidney-embedded mouse model, suggesting this pathway is central to the aggressive behavior.
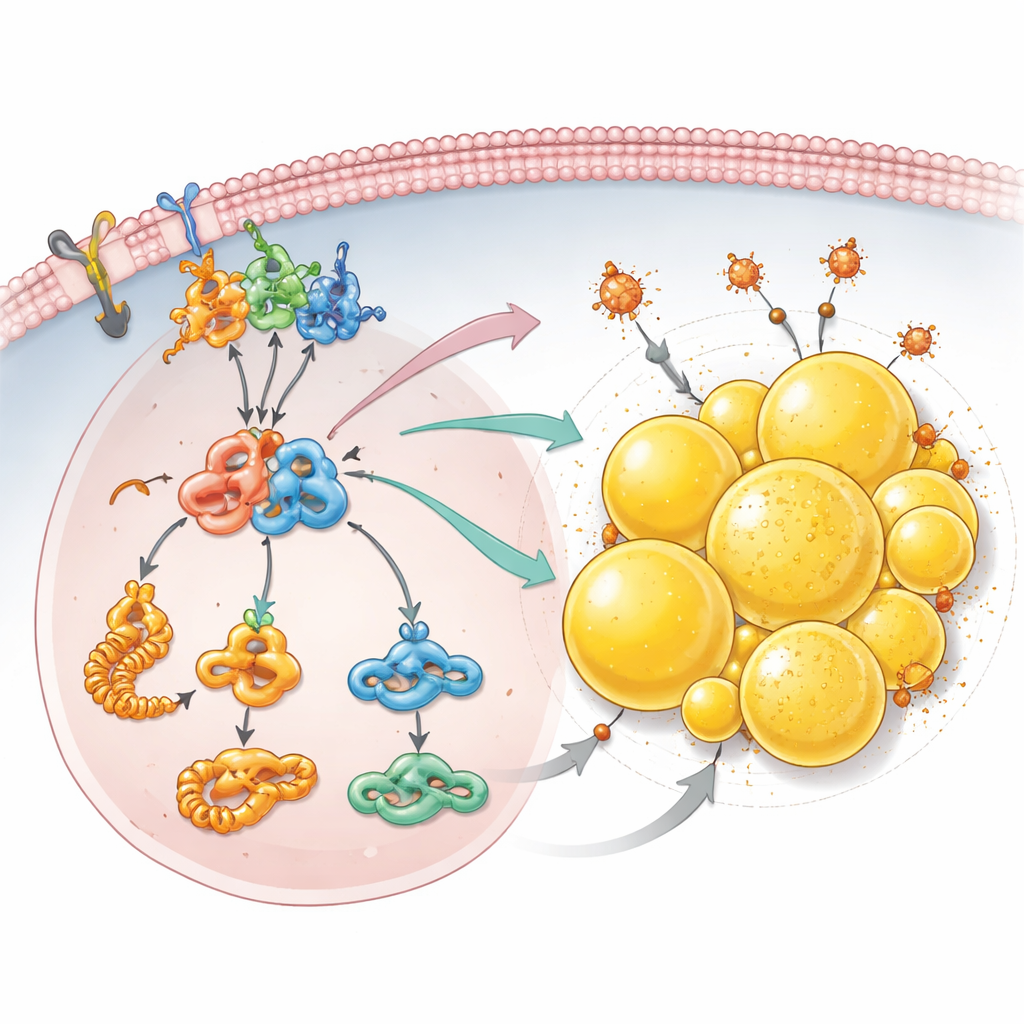
Fat storage as a shield against cell death
Beyond growth signals, the team discovered that PCDHGC3 loss rewires how cancer cells handle fats. Detailed protein surveys showed increased production of building blocks for fatty acids and higher levels of acetyl-CoA, a central fuel used to make lipids. Cells lacking PCDHGC3 built up many more lipid droplets—small fat-filled spheres inside the cell that are especially abundant in this type of kidney cancer. This accumulation depended on the mTOR–HIF2α axis and on PLIN2, a droplet-coating protein whose levels rose when PCDHGC3 was silenced. These droplets did more than just store excess fat: they helped the cells escape ferroptosis, a form of cell death driven by iron and damaged lipids. When PLIN2 was reduced, especially in cells already missing PCDHGC3, lipid droplets shrank and the cells became far more sensitive to a drug that triggers ferroptosis.
Turning a tumor’s survival trick into its weakness
Taken together, the study reveals a chain of events in which loss of PCDHGC3 at the cell surface activates focal adhesion kinase, switches on mTOR and HIF2α, boosts fat synthesis, and builds protective lipid droplets coated by PLIN2. This network not only fuels tumor growth and spread but also allows cancer cells to hide vulnerable fats away from destructive reactions that would otherwise kill them. The authors propose that targeting this newly defined PCDHGC3–mTOR–HIF2α–PLIN2 axis could offer a multi-pronged strategy for advanced clear cell kidney cancer: combining mTOR or HIF2α inhibitors with drugs that induce ferroptosis, and potentially blocking PLIN2, may turn the tumor’s own metabolic adaptation into an Achilles’ heel.
Citation: Celada, L., Cubiella, T., San-Juan-Guardado, J. et al. PCDHGC3 silencing promotes clear cell renal cell carcinoma metastasis via mTOR/HIF2α activation, lipid metabolism rewiring, and ferroptosis evasion. Cell Death Dis 17, 409 (2026). https://doi.org/10.1038/s41419-026-08643-y
Keywords: clear cell renal cell carcinoma, PCDHGC3, mTOR HIF2alpha pathway, lipid droplets, ferroptosis