Clear Sky Science · pl
Funkcje zależne i niezależne od transkrypcji izoform p53 u Drosophila w indukcji apoptozy i związanej z senescencją tumorogenezie
Dlaczego to badanie ma znaczenie
Nasze komórki nieustannie doświadczają uszkodzeń spowodowanych światłem słonecznym, chemikaliami i normalnym zużyciem. Białko o nazwie p53 jest znane z podejmowania decyzji, czy uszkodzona komórka powinna się naprawić, przestać się dzielić czy umrzeć. Gdy p53 działa wadliwie, często pojawiają się nowotwory. To badanie wykorzystuje muszkę owocową, Drosophila, by rozplątać, jak różne wersje p53 popychają komórki albo ku bezpiecznej autodestrukcji, albo ku niebezpiecznemu stanowi sprzyjającemu nowotworzeniu. Ponieważ p53 muszki i człowieka są zaskakująco podobne, wyniki te pomagają wyjaśnić, dlaczego niektóre odpowiedzi komórek na uszkodzenie zapobiegają rakowi, podczas gdy inne mogą go po cichu napędzać.
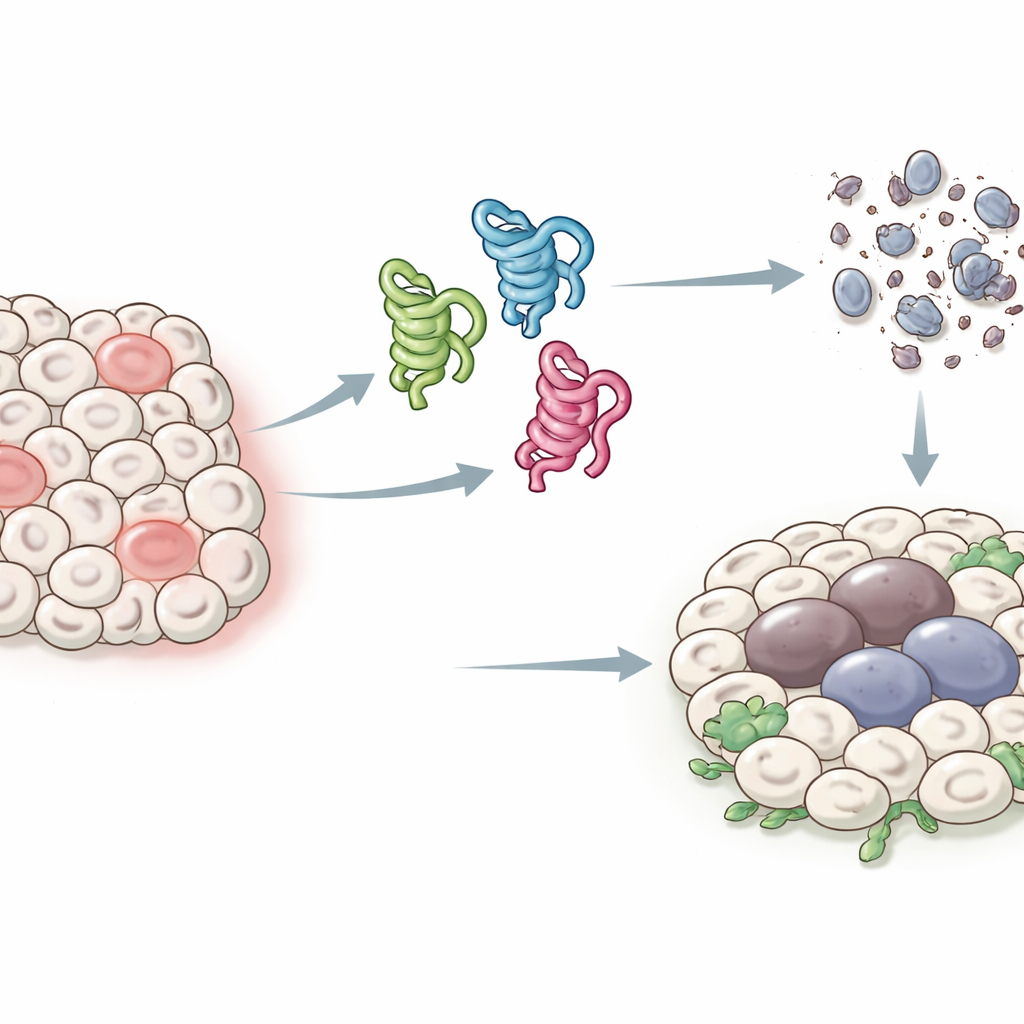
Różne oblicza białka-stróża
U ludzi gen p53 może dawać przynajmniej tuzin nieco różnych form białkowych, czyli izoform, co utrudnia przypisanie funkcji każdej z nich. Muszki mają prostszy układ: jeden gen p53, który produkuje zaledwie trzy główne izoformy, nazwane p53-A, p53-B i p53-E. Wszystkie trzy dzielą centralny region wiążący DNA, ale różnią się na końcach, co zmienia ich interakcje z innymi białkami. Autorzy wykorzystali ten uproszczony system, by ekspresyjnie wprowadzić każdą izoformę w określone tkanki muszki i sprawdzić, jak wpływają one na dwa kluczowe rezultaty po stresie lub uszkodzeniu DNA: programowaną śmierć komórki (apoptozę) oraz długotrwały stan zatrzymania przypominający starzenie komórkowe (senescencję), który może sprzyjać wzrostowi guza.
Kiedy szybkość podziału komórek zmienia wynik
Zespół najpierw sprawdził, czy cykl komórkowy, rytm podziałów komórkowych, zmienia sposób, w jaki każda izoforma p53 wywołuje apoptozę. Wymusili różne stany komórek: aktywnie dzielące się, zatrzymane w określonych fazach lub przełączone na wzór wzrostu omijający mitozę. Izoformy p53-A i p53-E zachowywały się jak ostrożni decydenci: silnie indukowały apoptozę tylko wtedy, gdy komórki aktywnie się dzieliły. Jeśli komórki były zatrzymane lub w specjalnym cyklu wzrostu, ich zdolność do wywoływania śmierci komórkowej w dużej mierze zanikała. Natomiast p53-B był znacznie bardziej bezkompromisowy. Wywoływał silną apoptozę niezależnie od tego, czy komórki się dzieliły, czy nie, zarówno w tarczkach imaginalnych (tkankach rozwojowych), jak i w dużych, nie dzielących się komórkach gruczołu ślinowego. To pokazało, że związek między p53 a cyklem komórkowym nie jest uniwersalny, lecz zależy od izoformy.
Droga na skróty do śmierci komórki omijająca geny
Klasyczny p53 działa jako czynnik transkrypcyjny: włącza geny takie jak reaper i hid, które uruchamiają maszynerię śmierci komórkowej. Autorzy potwierdzili, że p53-A i p53-E podążają tym scenariuszem. Gdy zablokowali te pro‑śmierci geny, apoptoza wywoływana przez p53-A i p53-E upadała. Ale p53-B złamał reguły. Nawet gdy główne geny proapoptotyczne zostały genetycznie usunięte, p53-B nadal skutecznie zabijał komórki. Szczegółowe eksperymenty wyjaśniły dlaczego: p53-B potrafi fizycznie wiązać się z Dronc, odpowiednikiem muszki inicjatorowej kaspazy, która znajduje się blisko początku kaskady śmierci. Za pomocą biochemicznych pull-downów i testu z rozdzielonym białkiem fluorescencyjnym uwidocznili specyficzną interakcję między p53-B a Dronc wewnątrz jąder komórkowych. Nawet wersja p53-B pozbawiona rdzenia wiążącego DNA potrafiła aktywować specjalnie zaprojektowany sensor Dronc i wywołać apoptozę, pod warunkiem obecności samego Dronc. To ujawnia bezpośrednią, niezależną od transkrypcji drogę od p53-B do maszynerii kaspaz.
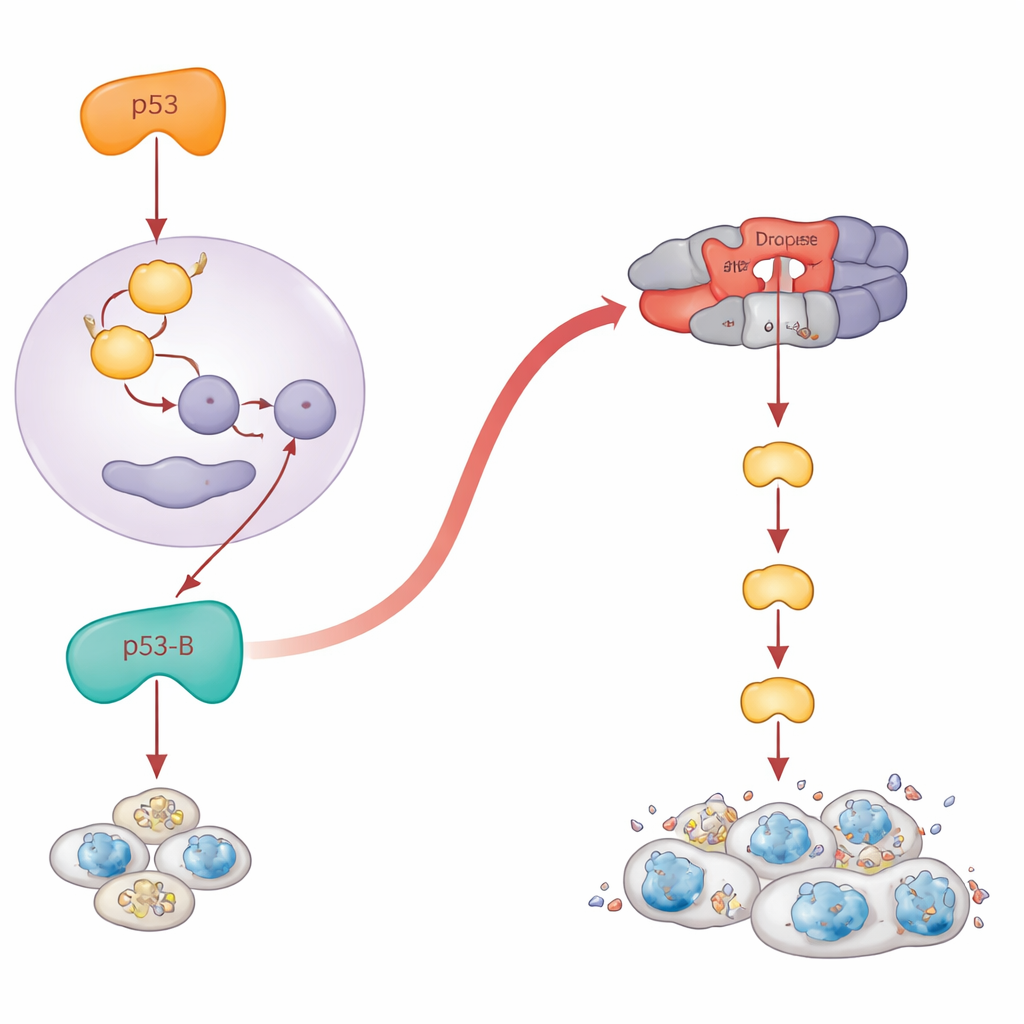
Kiedy nieudana śmierć przechodzi w niebezpieczny wzrost
Paradoksalnie, komórki, które nie umierają, mogą stać się długowieczne, zapalne i sprzyjające nowotworowi. Autorzy zapytali, co robi każda izoforma p53 w tkankach, gdzie apoptoza jest zablokowana. Ekspresowali izoformy w rozwijającym się skrzydle, zarówno w warunkach normalnych, jak i u muszek pozbawionych kluczowych genów śmierci. Gdy apoptoza była wyłączona, p53-A i p53-E przestały zmniejszać tkankę. Zamiast tego wytworzyły kieszonki komórek przypominających senescentne, które włączyły szlak stresowy zwany JNK i wydzielały sygnały wzrostowe. Te obszary rozszerzały się dramatycznie, tworząc przerostowe, przypominające guzy struktury wokół uszkodzonych komórek. Co godne uwagi, p53-B także napędzał tego rodzaju przerost, ale tylko wtedy, gdy apoptoza była zablokowana dalej w dół ścieżki, na poziomie Dronc; jeśli wczesne pro‑śmierci geny były wyłączone, p53-B nadal potrafił zabić komórki i nie obserwowano przerostu. W przypadku p53-A i p53-E włączenie JNK i wzrost nowotworopodobny wymagało ich zdolności wiązania DNA, podczas gdy p53-B mógł aktywować JNK i promować przerost związany z senescencją nawet bez wiązania DNA, co ponownie wskazuje na działania nieoparte na transkrypcji.
Czego muszka uczy nas o ludzkim p53
Następnie badacze wprowadzili ludzką izoformę p53 (pełnometrażowy TAp53α) do tkanek muszki. Ludzki p53 zachowywał się częściowo jak muszy p53-A: głównie zabijał komórki poprzez uruchamianie genów proapoptotycznych i potrzebował inicjatorowej kaspazy Dronc. Jego siła zabijania była osłabiona, ale nie zniesiona, gdy cykl komórkowy został zatrzymany, a wersja pozbawiona rdzenia wiążącego DNA potrafiła słabo aktywować bezpośrednio Dronc, dając posmak zachowanej, niezależnej od transkrypcji drogi. Była jednak ważna różnica: w tkankach pozbawionych apoptozy ludzki p53 nie wywoływał przerostowego wzrostu przypominającego guz. Zamiast tego silnie zatrzymywał cykl komórkowy, redukując replikację DNA i utrzymując kontrolę nad rozmiarem tkanki, podobnie jak jego dobrze znana rola jako hamulca podziałów komórek u ludzi.
Wnioski z szerokiej perspektywy
Ta praca pokazuje, że nie wszystkie izoformy p53 działają tak samo. Niektóre, jak musze p53-A i p53-E, polegają głównie na włączaniu lub wyłączaniu genów i reagują czułe na to, czy komórki się dzielą. Inne, jak p53-B, potrafią całkowicie pominąć kontrolę DNA i podłączyć się bezpośrednio do rdzenia maszynerii śmierci albo do szlaków stresowych poprzez bezpośrednie oddziaływania białko–białko. W zdrowych tkankach te złożone mechanizmy pomagają upewnić się, że uszkodzone komórki albo umierają w sposób uporządkowany, albo wchodzą w kontrolowany stan senescencji. Jednak gdy apoptoza jest zablokowana, te same aktywności p53 mogą sprzyjać przewlekłemu sygnalizowaniu stresu i przerostowi tkanek. Przez rozdzielenie tych zachowań w prostym organizmie badanie pomaga wyjaśnić, jak różne wersje p53 mogą w niektórych kontekstach hamować nowotwory, a w innych je promować, i podkreśla konkretne interakcje — takie jak bezpośrednie powiązanie między p53 a kaspazami — które mogą być ważnymi celami dla przyszłych terapii przeciwnowotworowych.
Cytowanie: Pérez-Aguilera, M., Ruiz-Losada, M., Gil Cortes, P. et al. Transcription-dependent and -independent functions of Drosophila p53 isoforms in the induction of apoptosis and senescence-associated tumorigenesis. Cell Death Dis 17, 367 (2026). https://doi.org/10.1038/s41419-026-08571-x
Słowa kluczowe: izoformy p53, apoptoza, senescencja komórkowa, model Drosophila, tumorogeneza