Clear Sky Science · en
Transcription-dependent and -independent functions of Drosophila p53 isoforms in the induction of apoptosis and senescence-associated tumorigenesis
Why this study matters
Our cells constantly face damage from sunlight, chemicals, and normal wear and tear. A protein called p53 is famous for deciding whether a damaged cell should repair itself, stop dividing, or die. When p53 goes wrong, cancers often arise. This study uses the fruit fly, Drosophila, to untangle how different versions of p53 push cells either toward safe self-destruction or toward a dangerous, tumor-promoting state. Because fly and human p53 are surprisingly similar, these findings help clarify why some cell responses to damage prevent cancer while others may quietly fuel it.
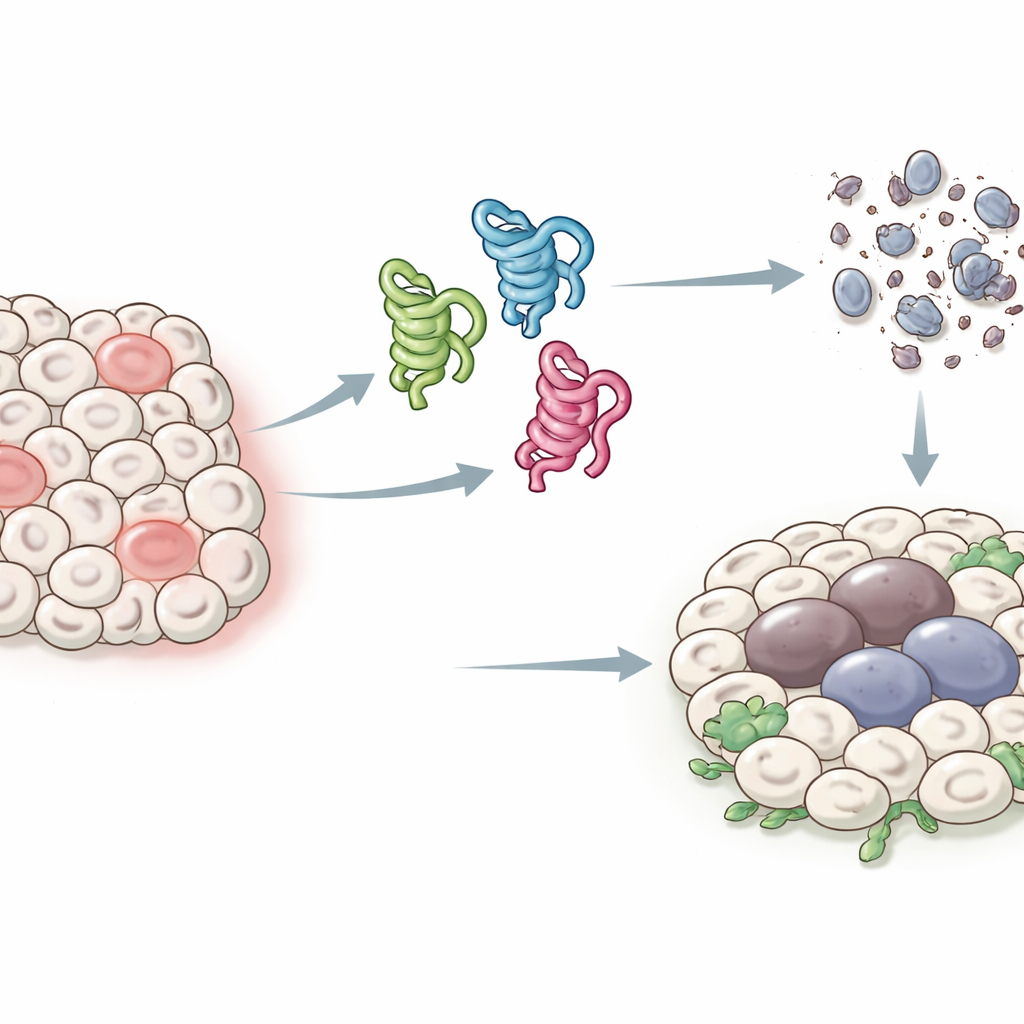
Different faces of a guardian protein
In humans, the p53 gene can produce at least a dozen slightly different protein forms, or isoforms, which has made it hard to pinpoint what each one does. Fruit flies have a simpler setup: a single p53 gene that makes just three main isoforms, called p53-A, p53-B, and p53-E. All three share a core region that can bind DNA, but they differ at their ends, which changes how they interact with other proteins. The authors took advantage of this streamlined system to express each isoform in specific fly tissues and ask how they influence two key outcomes after stress or DNA damage: programmed cell death (apoptosis) and a long-lasting arrest state resembling cellular aging (senescence) that can promote tumor growth.
When cell division speed changes the outcome
The team first tested whether the cell cycle, the rhythm of cell division, changes how each p53 isoform triggers apoptosis. They forced fly cells into different states: actively dividing, paused in particular phases, or switched into a growth pattern that skips mitosis. Isoforms p53-A and p53-E behaved like cautious decision-makers: they could strongly induce apoptosis only when cells were actively cycling. If the cells were arrested or in a special growth cycle, their ability to trigger cell death largely vanished. In contrast, p53-B was far more uncompromising. It drove robust apoptosis regardless of whether cells were dividing or not, in both imaginal discs (developing tissues) and in large, non-dividing salivary gland cells. This showed that the relationship between p53 and the cell cycle is not universal, but isoform-specific.
A shortcut to cell death that bypasses genes
Classic textbook p53 acts as a transcription factor: it turns on genes such as reaper and hid that set off the cell’s death machinery. The authors confirmed that p53-A and p53-E follow this script. When they blocked these pro-death genes, apoptosis caused by p53-A and p53-E collapsed. But p53-B broke the rules. Even when the main pro-apoptotic genes were genetically removed, p53-B still powerfully killed cells. Detailed experiments showed why: p53-B can physically bind to Dronc, the fly equivalent of an “initiator” caspase that sits near the top of the death cascade. Using biochemical pull-downs and a split-fluorescent protein assay, they visualized a specific interaction between p53-B and Dronc inside cell nuclei. Even a version of p53-B stripped of its DNA-binding core could still activate a specially designed Dronc sensor and trigger apoptosis, as long as Dronc itself was present. This reveals a direct, transcription-independent route from p53-B to the caspase machinery.
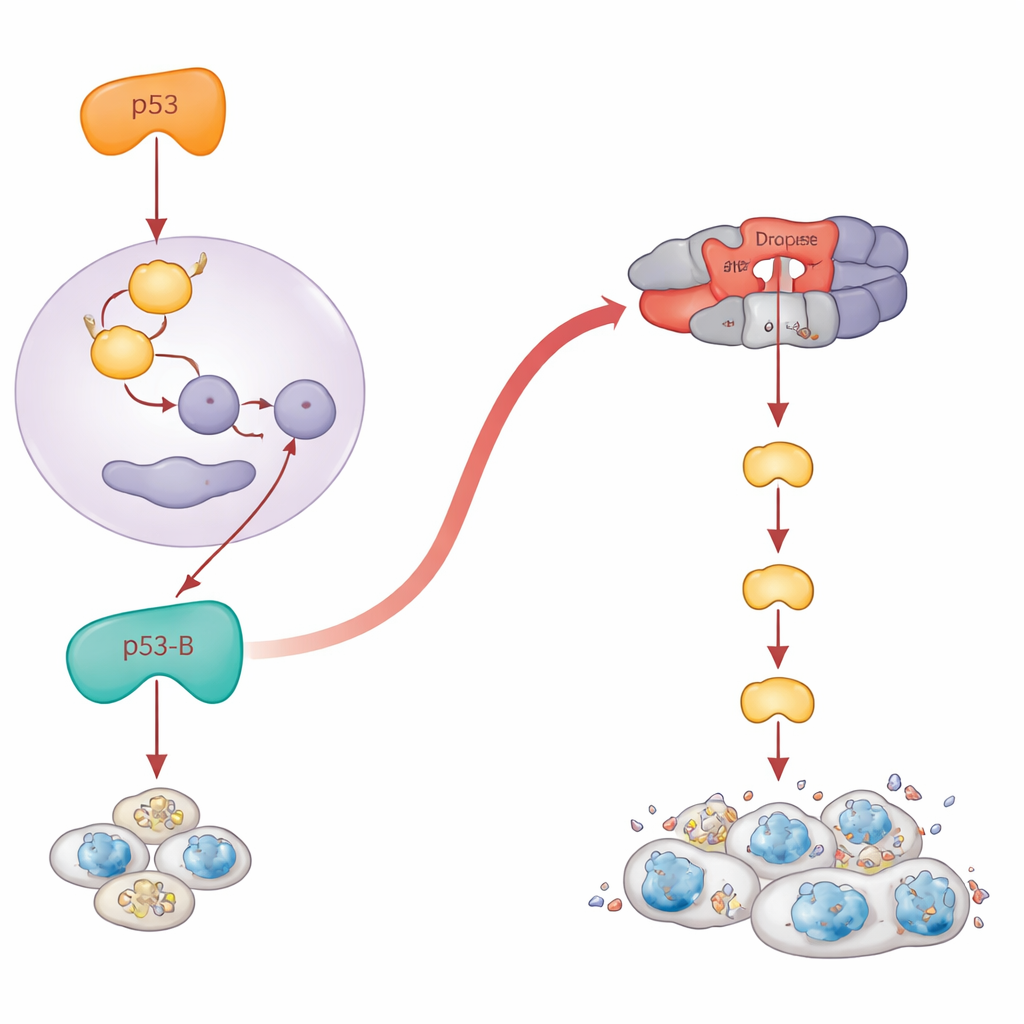
When failed death turns into dangerous growth
Paradoxically, cells that fail to die can become long-lived, inflamed, and tumor-promoting. The authors asked what each p53 isoform does in tissues where apoptosis is blocked. They expressed the isoforms in the developing wing, either in normal conditions or in flies lacking key death genes. When apoptosis was disabled, p53-A and p53-E no longer shrank the tissue. Instead, they produced pockets of senescent-like cells that switched on a stress pathway called JNK and secreted growth signals. These regions expanded dramatically, forming overgrown, tumor-like structures around the damaged cells. Strikingly, p53-B also drove this kind of overgrowth, but only when apoptosis was blocked downstream at the level of Dronc; if the earlier pro-death genes were disabled, p53-B still managed to kill cells and no overgrowth appeared. For p53-A and p53-E, turning on JNK and tumor growth required their DNA-binding ability, whereas p53-B could activate JNK and promote senescence-linked overgrowth even without binding DNA, pointing again to non-transcriptional actions.
What the fly teaches us about human p53
The researchers then introduced a human p53 isoform (the full-length TAp53α) into fly tissues. Human p53 behaved partly like fly p53-A: it mainly killed cells by turning on pro-apoptotic genes and needed the initiator caspase Dronc. Its killing power was reduced, but not abolished, when the cell cycle was halted, and a version lacking the DNA-binding core could still weakly activate Dronc directly, hinting at a conserved transcription-independent route. However, there was an important difference: in apoptosis-deficient tissues, human p53 did not drive tumor-like overgrowth. Instead, it strongly arrested the cell cycle, reducing DNA replication and keeping tissue size in check, much like its well-known role as a brake on human cell division.
Big-picture takeaways
This work shows that not all p53 isoforms act alike. Some, like fly p53-A and p53-E, rely mainly on switching genes on or off and respond sensitively to whether cells are dividing. Others, like p53-B, can bypass DNA control entirely and plug straight into the core death machinery, or into stress pathways, through direct protein–protein contacts. In healthy tissues, these layered mechanisms help ensure that damaged cells either die cleanly or enter a contained, senescent state. But when apoptosis is blocked, the very same p53 activities can foster chronic stress signaling and tissue overgrowth. By dissecting these behaviors in a simple animal, the study helps explain how different p53 versions can suppress tumors in some contexts yet promote them in others, and it highlights specific interactions—such as the direct link between p53 and caspases—that may be important targets for future cancer therapies.
Citation: Pérez-Aguilera, M., Ruiz-Losada, M., Gil Cortes, P. et al. Transcription-dependent and -independent functions of Drosophila p53 isoforms in the induction of apoptosis and senescence-associated tumorigenesis. Cell Death Dis 17, 367 (2026). https://doi.org/10.1038/s41419-026-08571-x
Keywords: p53 isoforms, apoptosis, cell senescence, Drosophila models, tumorigenesis