Clear Sky Science · de
Transkriptionsabhängige und -unabhängige Funktionen von Drosophila-p53-Isoformen bei der Induktion von Apoptose und seneszenzassoziierter Tumorentstehung
Warum diese Studie wichtig ist
Unsere Zellen sind ständig Schäden durch Sonnenlicht, Chemikalien und normalen Verschleiß ausgesetzt. Ein Protein namens p53 ist bekannt dafür, darüber zu entscheiden, ob eine geschädigte Zelle sich reparieren, die Teilung einstellen oder sterben soll. Geht p53 fehl, entsteht häufig Krebs. Diese Studie nutzt die Fruchtfliege Drosophila, um zu klären, wie verschiedene Varianten von p53 Zellen entweder in Richtung sicherer Selbstzerstörung oder in einen gefährlichen, tumorfördernden Zustand lenken. Da Fliegen- und menschliches p53 überraschend ähnlich sind, helfen diese Ergebnisse zu verstehen, warum einige Zellantworten auf Schäden Krebs verhindern, während andere ihn möglicherweise stillschweigend fördern.
Verschiedene Gesichter eines Schutzproteins
Beim Menschen kann das p53-Gen mindestens ein Dutzend leicht unterschiedlicher Proteinformen oder Isoformen produzieren, was es schwierig macht, die Funktion jeder einzelnen zuzuordnen. Fruchtfliegen haben ein einfacheres System: ein einzelnes p53-Gen, das nur drei Hauptisoformen erzeugt, genannt p53-A, p53-B und p53-E. Alle drei teilen eine Kernregion, die an DNA binden kann, unterscheiden sich jedoch an den Enden, was ihre Interaktionen mit anderen Proteinen verändert. Die Autoren nutzten dieses vereinfachte System, um jede Isoform in spezifischen Fliegengeweben zu exprimieren und zu untersuchen, wie sie zwei zentrale Folgen von Stress oder DNA-Schäden beeinflussen: den programmierten Zelltod (Apoptose) und einen langanhaltenden Stillstands-Zustand, der der Zellalterung (Seneszenz) ähnelt und Tumorwachstum fördern kann.
Wenn die Zellteilungsrate das Ergebnis verändert
Das Team prüfte zunächst, ob der Zellzyklus, der Rhythmus der Zellteilung, beeinflusst, wie jede p53-Isoform Apoptose auslöst. Sie zwangen Flienzellen in unterschiedliche Zustände: aktiv teilend, in bestimmten Phasen angehalten oder in ein Wachstumsprogramm geschaltet, das die Mitose überspringt. Die Isoformen p53-A und p53-E verhielten sich wie vorsichtige Entscheidungsträger: Sie konnten Apoptose nur stark induzieren, wenn die Zellen aktiv teilten. Waren die Zellen angehalten oder in einem speziellen Wachstumszyklus, verschwand ihre Fähigkeit, den Zelltod auszulösen, weitgehend. Im Gegensatz dazu war p53-B deutlich kompromissloser. Es löste robuste Apoptose unabhängig davon aus, ob die Zellen teilten oder nicht, sowohl in Imaginalscheiben (Entwicklungsgewebe) als auch in großen, nicht teilenden Speicheldrüsenzellen. Das zeigt, dass die Beziehung zwischen p53 und dem Zellzyklus nicht allgemein, sondern isoformspezifisch ist.
Eine Abkürzung zum Zelltod, die Gene umgeht
Das klassische p53 wirkt als Transkriptionsfaktor: Es schaltet Gene wie reaper und hid ein, die die Todesmaschinerie der Zelle in Gang setzen. Die Autoren bestätigten, dass p53-A und p53-E diesem Skript folgen. Wenn sie diese pro‑apoptotischen Gene blockierten, brach die durch p53-A und p53-E ausgelöste Apoptose zusammen. Aber p53-B brach die Regeln. Selbst wenn die wichtigsten pro‑apoptotischen Gene genetisch entfernt wurden, tötete p53-B Zellen weiterhin wirksam. Detaillierte Experimente zeigten den Grund: p53-B kann physisch an Dronc binden, das Fliegenäquivalent einer „Initiator“-Caspase, die nahe der Spitze der Todeskaskade sitzt. Mit biochemischen Pull-downs und einem Split‑Fluoreszenz‑Assay visualisierten sie eine spezifische Interaktion zwischen p53-B und Dronc innerhalb von Zellkernen. Sogar eine Version von p53-B, der der DNA-bindende Kern fehlt, konnte noch einen speziell konstruierten Dronc‑Sensor aktivieren und Apoptose auslösen, solange Dronc selbst vorhanden war. Das offenbart einen direkten, transkriptionsunabhängigen Weg von p53-B zur Caspase‑Maschinerie.
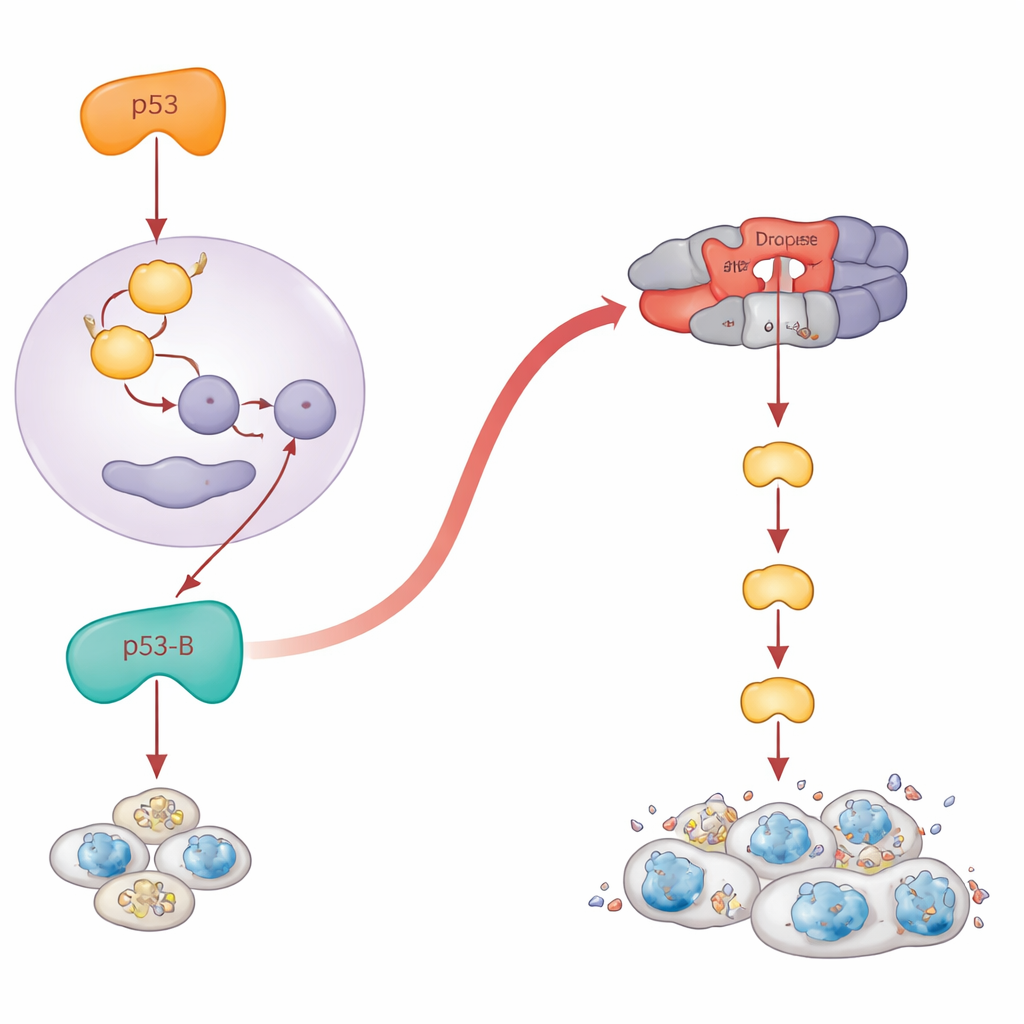
Wenn misslungener Tod zu gefährlichem Wachstum wird
Paradoxerweise können Zellen, denen das Sterben nicht gelingt, langlebig, entzündet und tumorfördernd werden. Die Autoren untersuchten, was jede p53-Isoform in Geweben bewirkt, in denen Apoptose blockiert ist. Sie exprimierten die Isoformen im sich entwickelnden Flügel, sowohl unter normalen Bedingungen als auch in Fliegen, denen wichtige Todesgene fehlen. War die Apoptose deaktiviert, verkleinerten p53-A und p53-E das Gewebe nicht mehr. Stattdessen erzeugten sie Ansammlungen seneszenzähnlicher Zellen, die einen Stressweg namens JNK aktivierten und Wachstumsfaktoren absonderten. Diese Regionen weiteten sich dramatisch aus und bildeten überwachsene, tumorähnliche Strukturen um die beschädigten Zellen. Auffällig war, dass p53-B ebenfalls dieses Überwachsungsverhalten antrieb, jedoch nur, wenn die Apoptose weiter stromabwärts auf der Ebene von Dronc blockiert war; waren die früheren pro‑apoptotischen Gene deaktiviert, gelang es p53-B weiterhin, Zellen zu töten, und es trat kein Überwachsen auf. Bei p53-A und p53-E erforderte die Aktivierung von JNK und Tumorwachstum ihre DNA‑Bindefähigkeit, während p53-B JNK aktivieren und seneszenz‑verknüpftes Überwachsen auch ohne DNA‑Bindung fördern konnte, was erneut auf transkriptionsunabhängige Aktionen hinweist.
Was die Fliege uns über menschliches p53 lehrt
Die Forscher führten dann eine menschliche p53‑Isoform (das volllängige TAp53α) in Fliegengewebe ein. Menschliches p53 verhielt sich teilweise wie das Fliegen‑p53‑A: Es tötete hauptsächlich Zellen durch das Einschalten pro‑apoptotischer Gene und benötigte die Initiator‑Caspase Dronc. Seine Tötungskraft war reduziert, aber nicht aufgehoben, wenn der Zellzyklus gestoppt wurde; eine Version ohne DNA‑bindenden Kern konnte Dronc noch schwach direkt aktivieren, was auf einen konservierten transkriptionsunabhängigen Weg hindeutet. Es gab jedoch einen wichtigen Unterschied: In apoptosis‑defizienten Geweben trieb menschliches p53 kein tumorähnliches Überwachsen voran. Stattdessen stoppte es stark den Zellzyklus, verringerte die DNA‑Replikation und hielt die Gewebegröße in Grenzen, ganz ähnlich seiner bekannten Rolle als Bremse der menschlichen Zellteilung.
Wesentliche Erkenntnisse
Diese Arbeit zeigt, dass nicht alle p53‑Isoformen gleich handeln. Manche, wie Fliegen‑p53‑A und p53‑E, verlassen sich hauptsächlich darauf, Gene an- oder auszuschalten, und reagieren sensibel darauf, ob Zellen teilen. Andere, wie p53‑B, können die DNA‑Kontrolle vollständig umgehen und direkt an die Kern‑Todesmaschinerie oder an Stresswege andocken, mittels direkter Protein‑Protein‑Interaktionen. In gesunden Geweben helfen diese geschichteten Mechanismen sicherzustellen, dass geschädigte Zellen entweder sauber sterben oder in einen eingegrenzten, seneszenten Zustand eintreten. Wenn die Apoptose blockiert ist, können gerade diese p53‑Aktivitäten jedoch chronische Stresssignale und Gewebeüberwachsen fördern. Durch die Zerlegung dieser Verhaltensweisen in einem einfachen Tier erklärt die Studie, wie verschiedene p53‑Varianten in manchen Kontexten Tumore unterdrücken, in anderen aber fördern können, und sie hebt spezifische Interaktionen hervor — etwa die direkte Verbindung zwischen p53 und Caspasen — die potenziell wichtige Ziele für künftige Krebstherapien sein könnten.
Zitation: Pérez-Aguilera, M., Ruiz-Losada, M., Gil Cortes, P. et al. Transcription-dependent and -independent functions of Drosophila p53 isoforms in the induction of apoptosis and senescence-associated tumorigenesis. Cell Death Dis 17, 367 (2026). https://doi.org/10.1038/s41419-026-08571-x
Schlüsselwörter: p53-Isoformen, Apoptose, Zellseneszenz, Drosophila-Modelle, Tumorentstehung