Clear Sky Science · fr
PHIP supprime NuRD pour permettre la croissance des cancers mutés du complexe SWI/SNF
Pourquoi cette étude compte pour le cancer
Les cancers surviennent souvent lorsque la machinerie cellulaire qui active ou réprime les gènes dysfonctionne. Cet article explore comment une protéine peu connue, appelée PHIP, aide certaines tumeurs infantiles et ovariennes très agressives à survivre, et pourquoi bloquer PHIP pourrait offrir une voie thérapeutique nouvelle et sélective pour des tumeurs qui ont aujourd’hui peu d’options efficaces.
Tiraillement autour du script de l’ADN
À l’intérieur de nos cellules, l’ADN s’enroule autour de protéines pour former la chromatine, et de larges machines moléculaires modulent constamment cette architecture pour ouvrir des gènes à l’expression ou les refermer. Un acteur majeur de « l’ouverture » est le complexe SWI/SNF, qui utilise l’énergie chimique pour desserrer l’ADN et permettre l’activité génique. Du côté de la « fermeture » se trouvent des complexes comme NuRD, qui compactent la chromatine et retirent des marques chimiques favorisant l’expression. Dans environ 20 % des cancers, y compris des tumeurs rares mais mortelles appelées rhabdoïdes et certains cancers ovariens apparentés, des composants centraux de SWI/SNF sont perdus, affaiblissant fortement le versant activateur de cet équilibre.
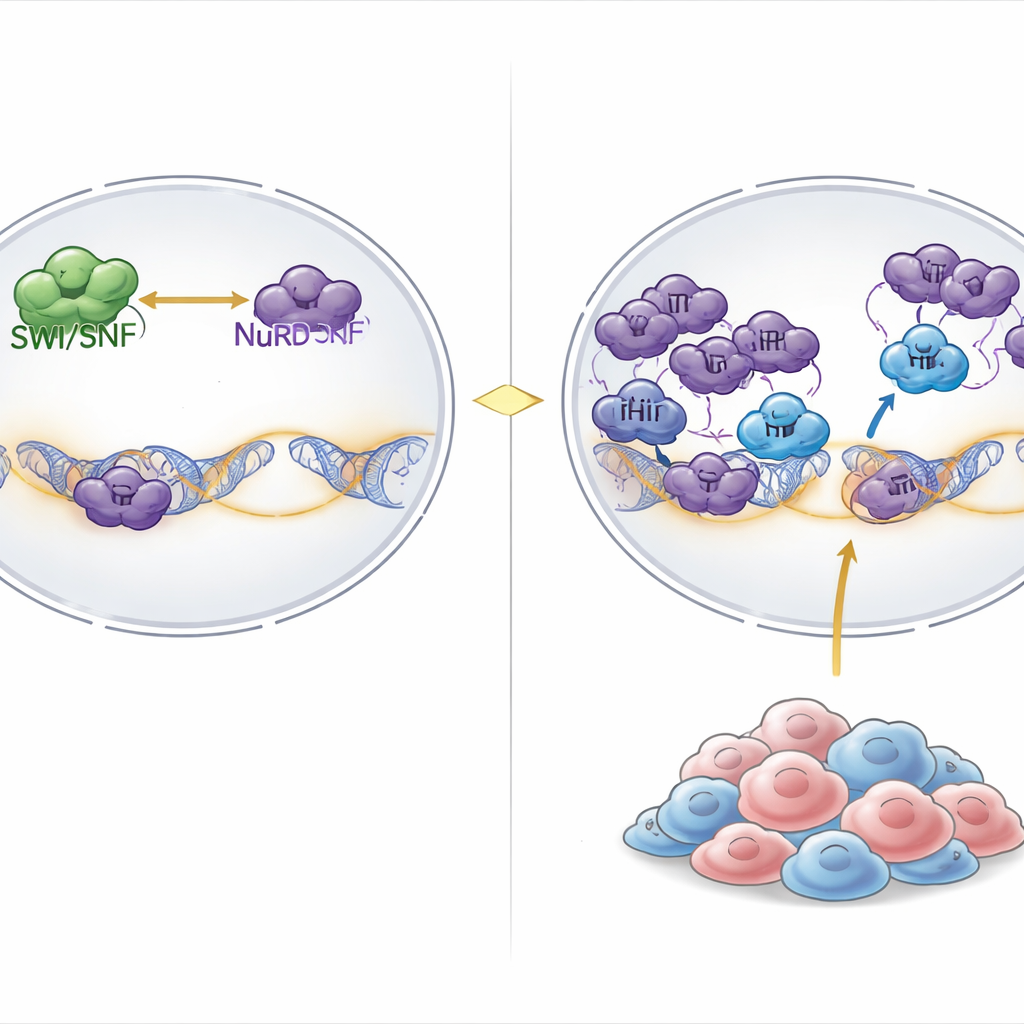
Détecter une faiblesse cachée dans les tumeurs mutées SWI/SNF
Les chercheurs ont analysé des résultats d’écrans CRISPR de perte de gène à l’échelle du génome réalisés sur plus d’un millier de lignées cellulaires cancéreuses. Ils ont cherché quels gènes sont spécifiquement essentiels dans les cancers présentant une perturbation étendue de SWI/SNF. Un candidat marquant fut PHIP, une protéine liant la chromatine auparavant associée à la réplication de l’ADN mais non clairement impliquée dans le contrôle transcriptionnel. Lorsque PHIP était désactivée, la croissance des cellules de tumeurs rhabdoïdes et ovariennes mutées de SWI/SNF chutait fortement, alors que la plupart des autres types de cancer toléraient la perte de PHIP. Introduire des défauts SWI/SNF dans des cellules normalement résistantes les rendait sensibles à la perte de PHIP, montrant que la dépendance à PHIP est une conséquence directe du dysfonctionnement de SWI/SNF plutôt qu’une question de type tissulaire.
Comment PHIP maintient les gènes de croissance activés
Pour comprendre la fonction de PHIP, l’équipe a cartographié ses sites d’ancrage sur la chromatine et mesuré les variations d’activité génique lorsque les niveaux de PHIP augmentent ou diminuent. PHIP se localisait près des sites de départ des gènes déjà marqués chimiquement comme actifs. La réduction de PHIP entraînait la baisse d’expression de nombreux de ces gènes, en particulier ceux qui pilotent la division cellulaire, tandis qu’une surexpression de PHIP produisait l’effet inverse. PHIP s’est également révélé crucial pour le maintien de marques d’acétylation spécifiques sur les histones — drapeaux chimiques signalant un état de chromatine ouvert et favorable à la transcription — à ces promoteurs. En l’absence de PHIP, l’acétylation décline et les gènes correspondants se taisent, cohérent avec le rôle de PHIP en tant que gardien local des régions géniques actives.
PHIP en tant que videur face à un complexe répressif
PHIP ne possède pas d’activité enzymatique propre, les auteurs ont donc cherché s’il agit en recrutant d’autres facteurs. Ils ont trouvé que PHIP amène un assemblage enzymatique nommé ligase ubiquitine CRL4 sur la chromatine. Cette ligase attache de petites étiquettes ubiquitine à des protéines cibles, changeant souvent leur comportement ou leur localisation. La spectrométrie de masse et des tests biochimiques ont montré que le complexe PHIP–CRL4 marque directement des éléments clés du complexe NuRD, en particulier la sous-unité de remodelage CHD4. Dans les cellules cancéreuses, PHIP était nécessaire pour que CRL4 interagisse avec NuRD sur l’ADN, conduisant à l’ubiquitination de NuRD et à son retrait des promoteurs. Quand PHIP disparaissait, NuRD s’accumulait aux sites de départ des gènes, les marques d’acétylation locales chutaient et les gènes voisins étaient silencieux. De manière importante, diminuer CHD4 pouvait inverser la perte d’acétylation causée par l’inactivation de PHIP, attribuant l’effet à l’activité de remodelage de NuRD.
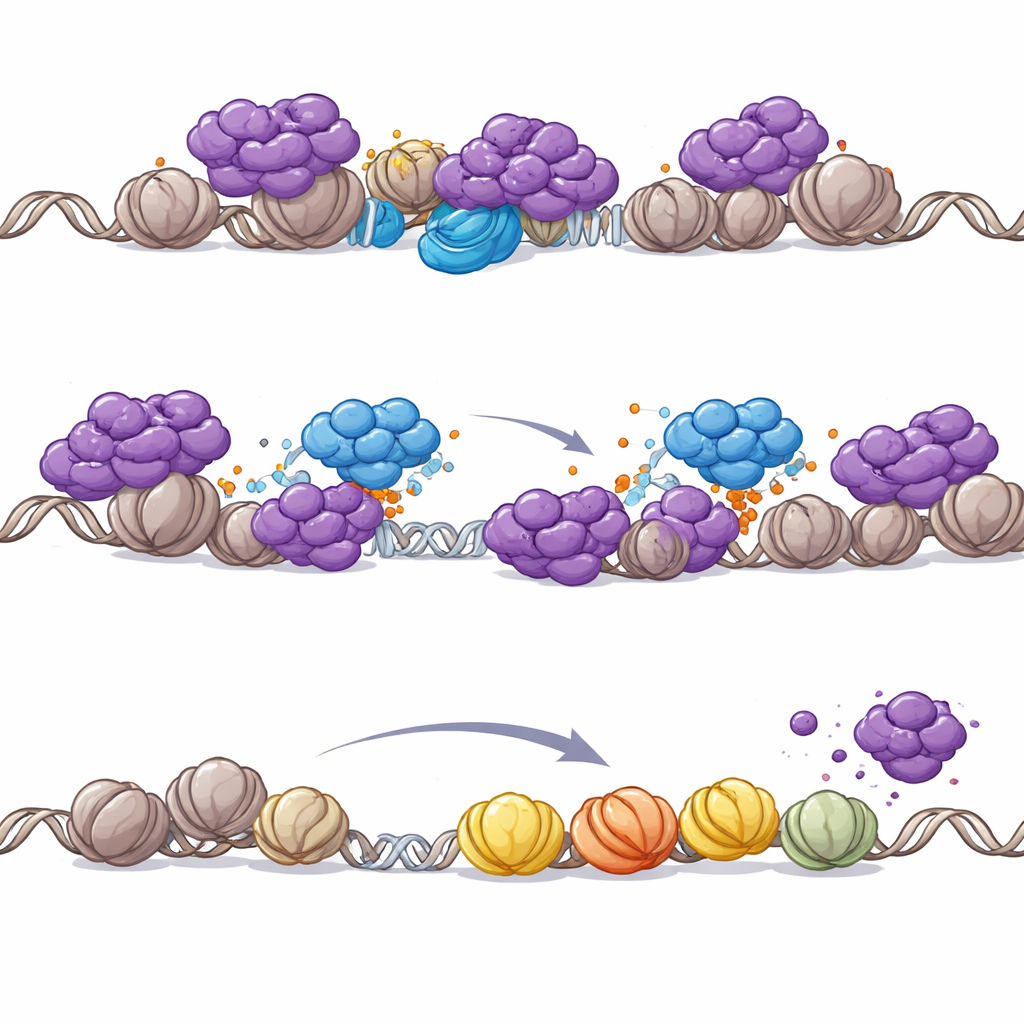
Pourquoi PHIP devient critique lorsque SWI/SNF est détruit
L’équipe a ensuite étudié pourquoi l’interaction PHIP–NuRD est si importante spécifiquement dans les cancers mutés de SWI/SNF. Restaurer une sous-unité manquante de SWI/SNF dans des cellules de tumeur rhabdoïde a modifié la localisation de NuRD sur le génome : NuRD s’est déplacé des promoteurs vers des régions enhancers réactivées par SWI/SNF. En l’absence de SWI/SNF, cependant, NuRD se redistribue aux promoteurs, menaçant une fermeture étendue des gènes nécessaires à la survie. Dans ce contexte vulnérable, l’éviction de NuRD médiée par PHIP devient essentielle pour maintenir l’expression génique pilotée par les promoteurs. Même dans des cellules autrement normales, dégrader chimiquement les moteurs centraux de SWI/SNF suffisait à déclencher le retrait dépendant de PHIP de NuRD de la chromatine, renforçant que le rôle de PHIP est étroitement lié à la perte de SWI/SNF.
Des cultures en laboratoire aux tumeurs dérivées de patients
Pour tester la pertinence au-delà des lignées cellulaires, les chercheurs ont utilisé des organoïdes tumoraux cérébraux dérivés de patients et des modèles murins de tumeurs rhabdoïdes. Dans des cultures tumorales tridimensionnelles issues de cancers infantiles présentant des défauts de SWI/SNF, la perturbation de PHIP réduisait nettement la fitness des cellules tumorales, tandis que des organoïdes d’un cancer cérébral apparenté sans mutation SWI/SNF étaient largement indemnes. Quand des organoïdes avec PHIP supprimé furent implantés dans le cerveau de souris, les animaux survécurent beaucoup plus longtemps que ceux porteurs de tumeurs témoins. Les tumeurs qui ont fini par récidiver dans le groupe PHIP knockout avaient partiellement restauré les niveaux de PHIP, indiquant que les cellules cancéreuses ayant échappé à la perte de PHIP étaient celles capables de repousser.
Ce que cela signifie pour les traitements futurs du cancer
Ce travail identifie PHIP comme un « garde du corps » moléculaire qui protège l’activité génique dans les cancers où la puissance activateur de SWI/SNF est affaiblie. En recrutant une ligase pour expulser le complexe répressif NuRD des promoteurs critiques, PHIP permet aux tumeurs mutées de SWI/SNF de maintenir l’expression de gènes essentiels à la croissance. Parce que la plupart des autres types de cancer — et vraisemblablement de nombreuses cellules normales — ne dépendent pas autant de PHIP, des médicaments inhibant sa poche de liaison spécifique pourraient, en principe, frapper plus durement les tumeurs rhabdoïdes et apparentées que les tissus sains. Bien que de tels traitements restent à développer et à tester cliniquement, l’étude fournit une justification mécanistique claire pour cibler PHIP comme nouvelle stratégie contre certains des cancers mutés SWI/SNF les plus difficiles.
Citation: Malone, H.A., Myers, J.A., Gruss, E.G. et al. PHIP suppresses NuRD to enable the growth of SWI/SNF-mutant cancers. Nat Commun 17, 2877 (2026). https://doi.org/10.1038/s41467-026-70699-3
Mots-clés: thérapie épigénétique contre le cancer, mutations SWI/SNF, complexe NuRD, remodelage de la chromatine, tumeurs rhabdoïdes