Clear Sky Science · en
PHIP suppresses NuRD to enable the growth of SWI/SNF-mutant cancers
Why this study matters for cancer
Cancers often arise when the cellular machinery that switches genes on and off goes awry. This paper explores how a lesser-known protein called PHIP helps certain highly aggressive childhood and ovarian cancers survive, and why blocking PHIP could offer a new, selective way to treat tumors that currently have few good options.
The tug-of-war over the DNA script
Inside our cells, DNA is wrapped around protein spools to form chromatin, and large molecular machines constantly nudge this structure to either open genes for use or close them down. One major “opening” machine is the SWI/SNF complex, which uses chemical energy to loosen DNA and allow gene activity. On the “closing” side are complexes like NuRD, which tighten chromatin and remove chemical marks that favor gene expression. In about 20% of cancers, including rare but lethal tumors called rhabdoid tumors and related ovarian cancers, core pieces of SWI/SNF are lost, severely weakening the gene-activating side of this balance.
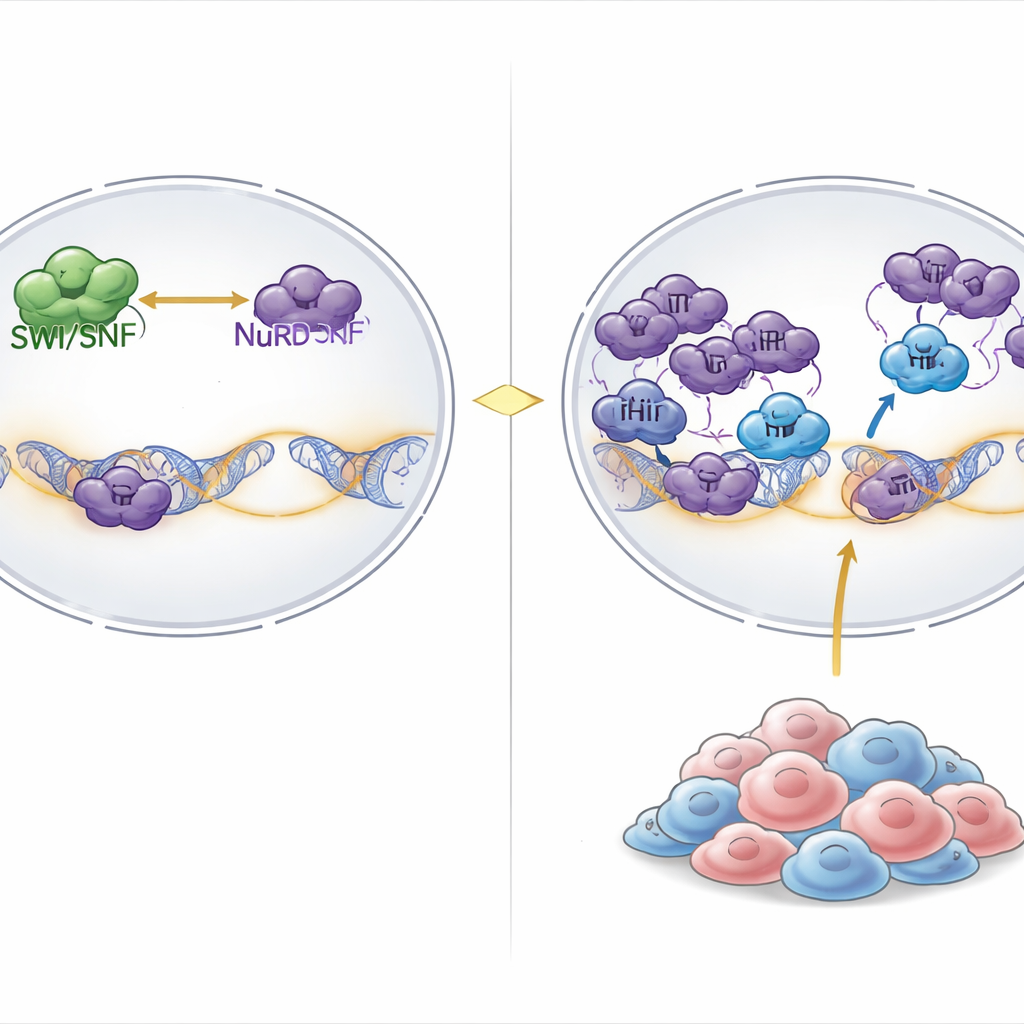
Finding a hidden weakness in SWI/SNF-mutant tumors
The researchers combed through results from genome-wide CRISPR gene knockout screens in more than a thousand cancer cell lines. They asked which genes were uniquely essential in cancers with broad SWI/SNF disruption. A standout hit was PHIP, a chromatin-binding protein previously linked to DNA replication but not clearly tied to gene control. When PHIP was disabled, growth of SWI/SNF-mutant rhabdoid and ovarian cancer cells sharply declined, whereas most other cancer types tolerated PHIP loss. Introducing SWI/SNF defects into normally resistant cells made them newly sensitive to losing PHIP, showing that PHIP dependence is a direct consequence of SWI/SNF malfunction rather than tissue type.
How PHIP keeps growth genes switched on
To understand what PHIP does, the team mapped where it sits on chromatin and how gene activity changes when PHIP levels rise or fall. PHIP clustered near gene start sites that are already chemically marked as active. Reducing PHIP caused many of these genes to be turned down, especially those that drive cell division, while boosting PHIP had the opposite effect. PHIP also proved crucial for maintaining specific acetyl marks on histone proteins—chemical flags that signal an open, transcription-friendly chromatin state—at these promoters. Without PHIP, acetylation faded and the corresponding genes quieted, consistent with PHIP acting as a local guardian of active gene regions.
PHIP as a bouncer for a repressive complex
PHIP lacks its own enzyme activity, so the authors asked whether it works by recruiting other factors. They found that PHIP brings an enzyme assembly called the CRL4 ubiquitin ligase onto chromatin. This ligase attaches small ubiquitin tags to target proteins, often changing their behavior or location. Mass spectrometry and biochemical tests revealed that PHIP–CRL4 directly tags key parts of the NuRD complex, particularly the CHD4 remodeling subunit. In cancer cells, PHIP was required for CRL4 to engage NuRD on DNA, leading to NuRD ubiquitination and its removal from promoters. When PHIP was lost, NuRD piled up at gene start sites, local acetyl marks dropped, and nearby genes were silenced. Importantly, dialing down CHD4 could reverse the acetylation loss caused by PHIP inactivation, pinning the effect on NuRD’s remodeling activity.
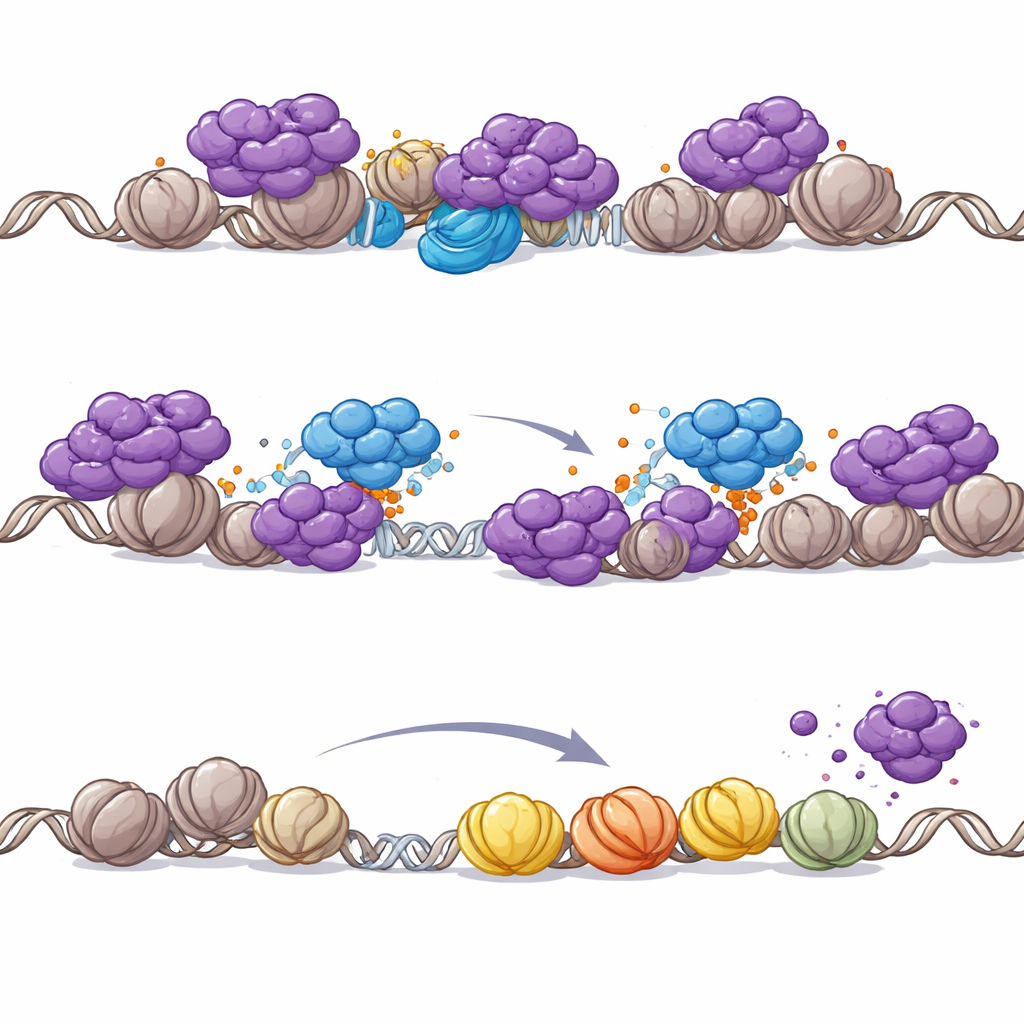
Why PHIP becomes critical when SWI/SNF is broken
The team then explored why this PHIP–NuRD interaction matters so much specifically in SWI/SNF-mutant cancers. Restoring a missing SWI/SNF subunit in rhabdoid tumor cells reshaped where NuRD sits on the genome: NuRD shifted away from promoters and toward enhancer regions that were reactivated by SWI/SNF. When SWI/SNF is absent, however, NuRD redistributes to promoters, threatening widespread shutdown of genes needed for survival. In this vulnerable setting, PHIP-mediated eviction of NuRD becomes essential to keep promoter-driven gene expression alive. Even in otherwise normal cells, chemically degrading SWI/SNF’s core engines was enough to trigger PHIP-dependent removal of NuRD from chromatin, reinforcing that PHIP’s role is tightly coupled to SWI/SNF loss.
From lab dishes to patient-derived tumors
To test relevance beyond cell lines, the researchers turned to patient-derived brain tumor organoids and mouse models of rhabdoid tumors. In three-dimensional tumor cultures grown from children’s cancers with SWI/SNF defects, disrupting PHIP markedly reduced the fitness of tumor cells, while organoids from a related brain cancer lacking SWI/SNF mutations were largely unaffected. When organoids with PHIP knocked out were implanted into the brains of mice, animals survived much longer than those harboring control tumors. Tumors that eventually relapsed in the PHIP knockout group had partially restored PHIP levels, indicating that cancer cells that escaped PHIP loss were the ones able to regrow.
What this means for future cancer treatments
This work reveals PHIP as a molecular “bodyguard” that protects gene activity in cancers where SWI/SNF’s usual activating power is crippled. By recruiting a tagging enzyme to kick the repressive NuRD complex off critical promoters, PHIP enables SWI/SNF-mutant tumors to keep key growth genes on. Because most other cancer types—and presumably many normal cells—do not rely as heavily on PHIP, drugs that inhibit its unique binding pocket could, in principle, hit vulnerable rhabdoid and related tumors much harder than healthy tissue. While such treatments remain to be developed and clinically tested, the study maps out a clear mechanistic rationale for targeting PHIP as a new strategy against some of the most challenging SWI/SNF-mutant cancers.
Citation: Malone, H.A., Myers, J.A., Gruss, E.G. et al. PHIP suppresses NuRD to enable the growth of SWI/SNF-mutant cancers. Nat Commun 17, 2877 (2026). https://doi.org/10.1038/s41467-026-70699-3
Keywords: epigenetic cancer therapy, SWI/SNF mutations, NuRD complex, chromatin remodeling, rhabdoid tumors