Clear Sky Science · de
PHIP unterdrückt NuRD, um das Wachstum von SWI/SNF-mutierten Krebsen zu ermöglichen
Warum diese Studie für Krebs wichtig ist
Krebs entsteht oft, wenn die zellulären Mechanismen, die Gene an- und ausschalten, fehlreguliert werden. Dieser Artikel untersucht, wie ein weniger bekanntes Protein namens PHIP bestimmten hochaggressiven Kinder- und Eierstockkrebsen das Überleben ermöglicht und warum die Blockade von PHIP einen neuen, selektiven Behandlungsansatz für Tumoren bieten könnte, für die es derzeit nur wenige gute Optionen gibt.
Der Tauziehen um das DNA-Skript
In unseren Zellen ist DNA um Proteinkerne gewickelt und große molekulare Maschinerien justieren konstant diese Struktur, um Gene entweder zu öffnen oder zu verschließen. Einer der wichtigsten „Öffner“ ist der SWI/SNF-Komplex, der chemische Energie nutzt, um DNA zu lockern und Genaktivität zu ermöglichen. Auf der „Schließer“-Seite stehen Komplexe wie NuRD, die Chromatin verdichten und chemische Markierungen entfernen, die für Genexpression förderlich sind. Bei etwa 20 % der Krebserkrankungen — darunter seltene, aber tödliche Rhabdoidtumoren und verwandte Eierstocktumoren — gehen zentrale Bausteine von SWI/SNF verloren, wodurch die genaktivierende Seite dieses Gleichgewichts stark geschwächt wird.
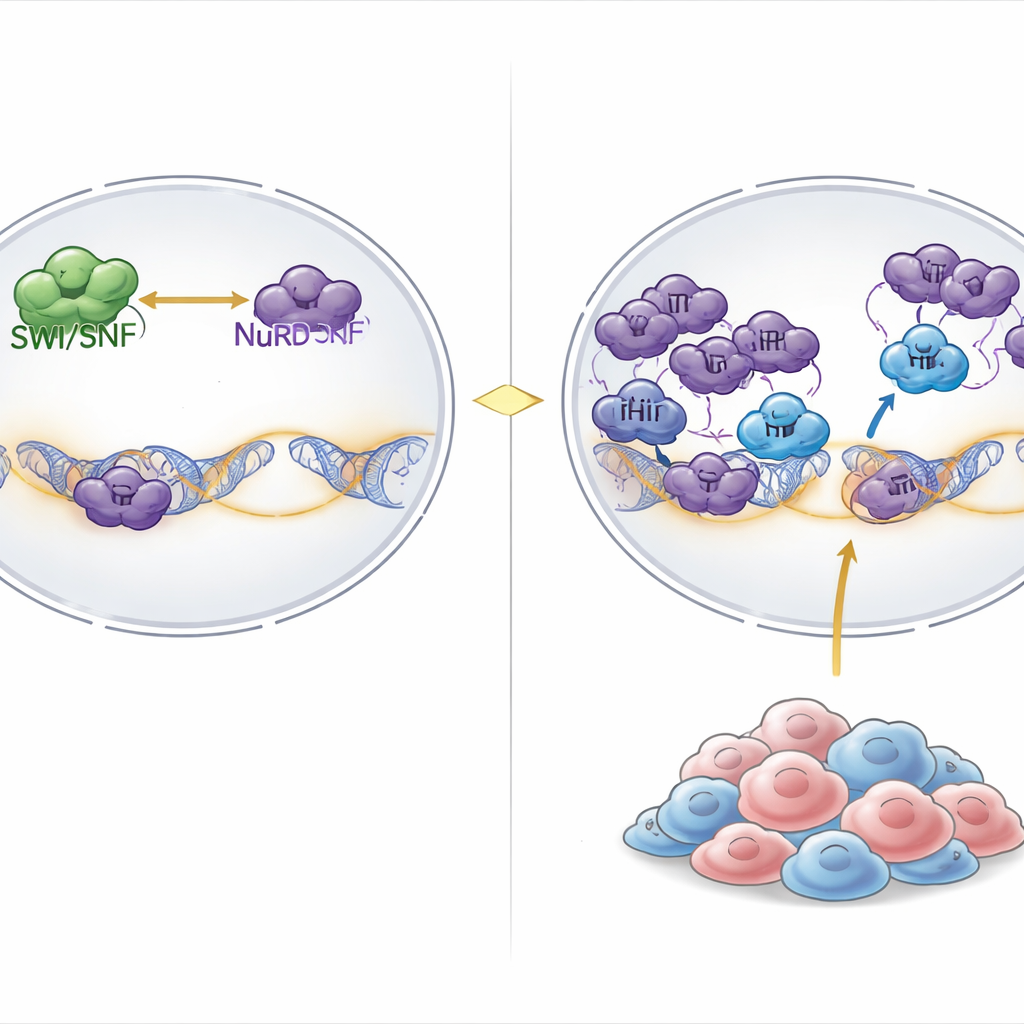
Eine verborgene Schwachstelle in SWI/SNF-mutierten Tumoren finden
Die Forschenden werteten Ergebnisse genomweiter CRISPR-Knockout-Screens in mehr als tausend Krebszelllinien aus. Sie fragten, welche Gene in Krebsen mit weitreichender SWI/SNF-Störung einzigartig essentiell sind. Ein herausragender Treffer war PHIP, ein chromatinbindendes Protein, das zuvor mit DNA-Replikation in Verbindung gebracht wurde, aber nicht klar mit Genregulation. Wurde PHIP ausgeschaltet, ging das Wachstum von SWI/SNF-mutierten Rhabdoid- und Eierstockkrebszellen deutlich zurück, während die meisten anderen Krebsarten den Verlust von PHIP tolerierten. Das Einführen von SWI/SNF-Defekten in normalerweise resistente Zellen machte diese neu empfindlich gegenüber PHIP-Verlust, was zeigt, dass die Abhängigkeit von PHIP eine direkte Folge der SWI/SNF-Fehlfunktion und nicht des Gewebetyps ist.
Wie PHIP Wachstums-Gene eingeschaltet hält
Um zu verstehen, was PHIP bewirkt, kartierte das Team seine Positionen auf dem Chromatin und wie sich die Genaktivität ändert, wenn PHIP-Level steigen oder fallen. PHIP gruppierte sich in der Nähe von Genstartstellen, die bereits chemisch als aktiv markiert waren. Eine Reduktion von PHIP führte dazu, dass viele dieser Gene herunterreguliert wurden, besonders solche, die die Zellteilung antreiben, während eine Erhöhung von PHIP den gegenteiligen Effekt hatte. PHIP erwies sich außerdem als entscheidend für die Aufrechterhaltung spezifischer Acetylierungen an Histonproteinen — chemische Signale, die einen offenen, transkriptionsfreundlichen Chromatinzustand anzeigen — an diesen Promotoren. Ohne PHIP verblassten die Acetylierungen und die entsprechenden Gene verstummten, was mit PHIP als lokalem Wächter aktiver Genregionen übereinstimmt.
PHIP als Türsteher für einen repressiven Komplex
PHIP besitzt keine eigene Enzymaktivität, daher untersuchten die Autoren, ob es durch Rekrutierung anderer Faktoren wirkt. Sie fanden heraus, dass PHIP eine Enzymassemblierung namens CRL4-Ubiquitin-Ligase auf das Chromatin bringt. Diese Ligase heftet kleine Ubiquitin-Tags an Zielproteine, was häufig deren Verhalten oder Lokalisation verändert. Massenspektrometrie und biochemische Tests zeigten, dass PHIP–CRL4 direkt Schlüsselbestandteile des NuRD-Komplexes markiert, insbesondere die CHD4-Remodelling-Untereinheit. In Krebszellen war PHIP erforderlich, damit CRL4 NuRD an der DNA anbindet, was zu NuRD-Ubiquitinierung und dessen Entfernung von Promotoren führte. Wurde PHIP verloren, sammelte sich NuRD an Genstartstellen an, lokale Acetylmarken sanken und nahegelegene Gene wurden stillgelegt. Wichtig war, dass die Herunterregelung von CHD4 den durch PHIP-Inaktivierung verursachten Acetylverlust rückgängig machen konnte, womit die Wirkung auf NuRDs Remodelling-Aktivität zurückgeführt wurde.
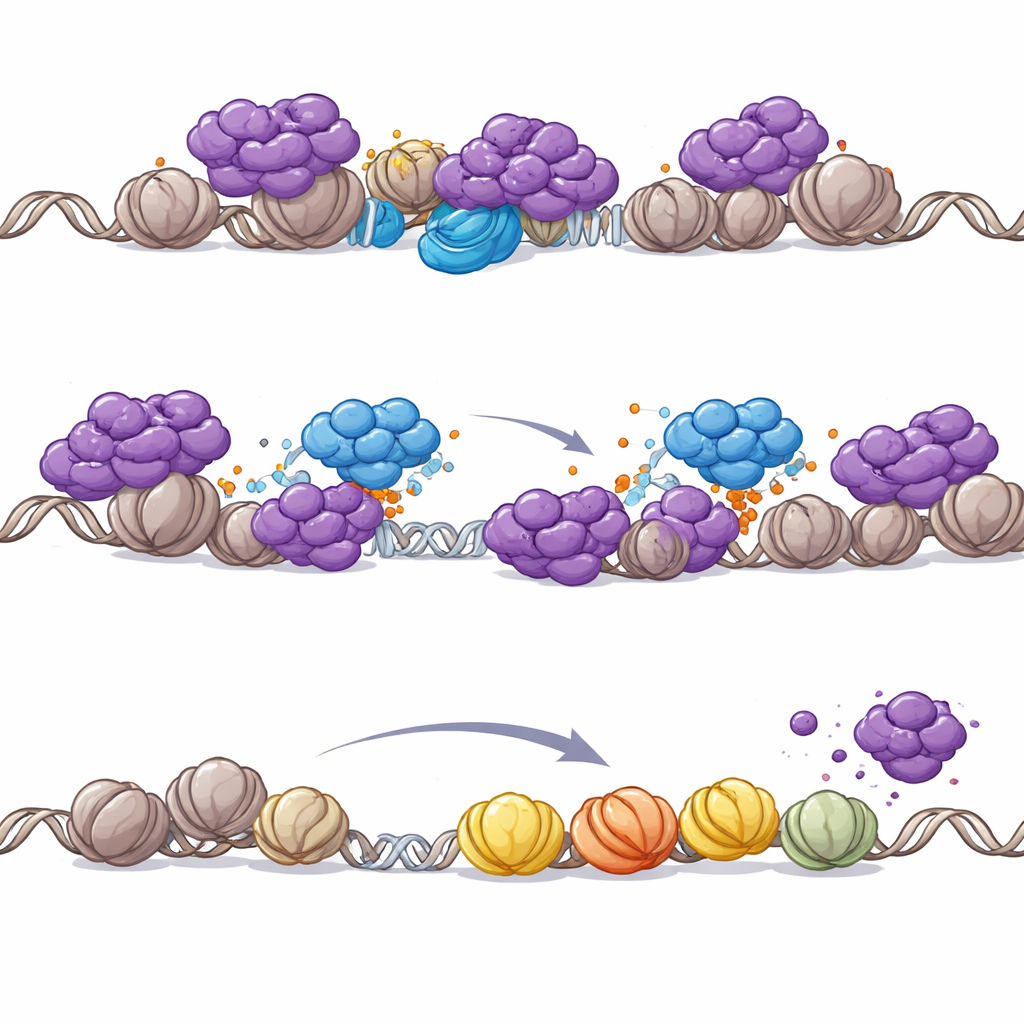
Warum PHIP kritisch wird, wenn SWI/SNF defekt ist
Das Team untersuchte dann, warum diese PHIP–NuRD-Interaktion gerade in SWI/SNF-mutierten Krebsen so wichtig ist. Die Wiederherstellung einer fehlenden SWI/SNF-Untereinheit in Rhabdoidtumorzellen veränderte die Genomposition von NuRD: NuRD verlagerte sich weg von Promotoren hin zu Enhancer-Regionen, die durch SWI/SNF reaktiviert wurden. Fehlt SWI/SNF jedoch, verteilt NuRD sich auf Promotoren, was eine weitreichende Abschaltung von Überlebensgenen drohen lässt. In diesem verletzlichen Zustand wird die PHIP-vermittelte Vertreibung von NuRD essenziell, um die promotorgetriebene Genexpression aufrechtzuerhalten. Selbst in ansonsten normalen Zellen reichte das chemische Abbau der zentralen SWI/SNF-Komponenten aus, um eine PHIP-abhängige Entfernung von NuRD vom Chromatin auszulösen, was unterstreicht, dass PHIPs Rolle eng mit dem Verlust von SWI/SNF verknüpft ist.
Vom Labor zu patientenabgeleiteten Tumoren
Um die Relevanz über Zelllinien hinaus zu prüfen, verwendeten die Forschenden patientenabgeleitete Hirntumor-Organoide und Mausmodelle von Rhabdoidtumoren. In dreidimensionalen Tumorkulturen aus Kinderkrebsen mit SWI/SNF-Defekten reduzierte die Störung von PHIP die Fitness der Tumorzellen deutlich, während Organoide eines verwandten Hirntumors ohne SWI/SNF-Mutationen weitgehend unbetroffen blieben. Wurden PHIP-knockout-Organoide in die Gehirne von Mäusen implantiert, überlebten die Tiere deutlich länger als solche mit Kontrolltumoren. Tumoren, die im PHIP-Knockout-Arm später wieder auftraten, zeigten teilweise wiederhergestellte PHIP-Level, was darauf hindeutet, dass Krebszellen, die dem PHIP-Verlust entkamen, die Wiederregeneration ermöglichten.
Was das für künftige Krebstherapien bedeutet
Diese Arbeit identifiziert PHIP als molekularen „Bodyguard“, der die Genaktivität in Tumoren schützt, in denen die übliche aktivierende Kraft von SWI/SNF geschwächt ist. Indem es eine Markierungsenzym-Rekrutierung einsetzt, um den repressiven NuRD-Komplex von kritischen Promotoren zu vertreiben, ermöglicht PHIP SWI/SNF-mutierten Tumoren, wichtige Wachstumsgene eingeschaltet zu halten. Da die meisten anderen Krebsarten — und vermutlich viele normale Zellen — nicht so stark von PHIP abhängen, könnten Wirkstoffe, die an seine einzigartige Bindetasche ansetzen, potenziell empfindliche Rhabdoid- und verwandte Tumoren deutlich stärker treffen als gesundes Gewebe. Solche Therapien müssen zwar noch entwickelt und klinisch getestet werden, doch die Studie liefert eine klare mechanistische Begründung dafür, PHIP als neuen Angriffsweg gegen einige der herausforderndsten SWI/SNF-mutierten Krebserkrankungen ins Visier zu nehmen.
Zitation: Malone, H.A., Myers, J.A., Gruss, E.G. et al. PHIP suppresses NuRD to enable the growth of SWI/SNF-mutant cancers. Nat Commun 17, 2877 (2026). https://doi.org/10.1038/s41467-026-70699-3
Schlüsselwörter: epigenetische Krebstherapie, SWI/SNF-Mutationen, NuRD-Komplex, Chromatin-Remodelling, Rhabdoidtumoren