Clear Sky Science · en
Profiling histone post-translational modifications to identify signatures of epigenetic drug response in T-cell acute lymphoblastic leukemia
Why this cancer study matters
Treatment for children and adults with T cell acute lymphoblastic leukemia has improved greatly, yet many patients still relapse or suffer serious side effects from intense chemotherapy. Doctors need better ways to predict which medicines will work for which patient. This study explores whether tiny chemical tags on DNA packaging proteins in leukemia cells can help forecast how those cells respond to modern cancer drugs that act on the cell’s epigenetic machinery.
The hidden code on DNA packaging
Inside each leukemia cell, DNA is wrapped around proteins called histones, forming a compact structure known as chromatin. Histones carry many small chemical tags that act like switches, helping to turn genes on or off without changing the underlying DNA sequence. Because these tags are reversible, they are attractive targets for drugs. Several such “epidrugs” are already used in blood cancers, but their role in T cell leukemia is still unclear, and doctors lack simple markers to predict who will benefit from them.
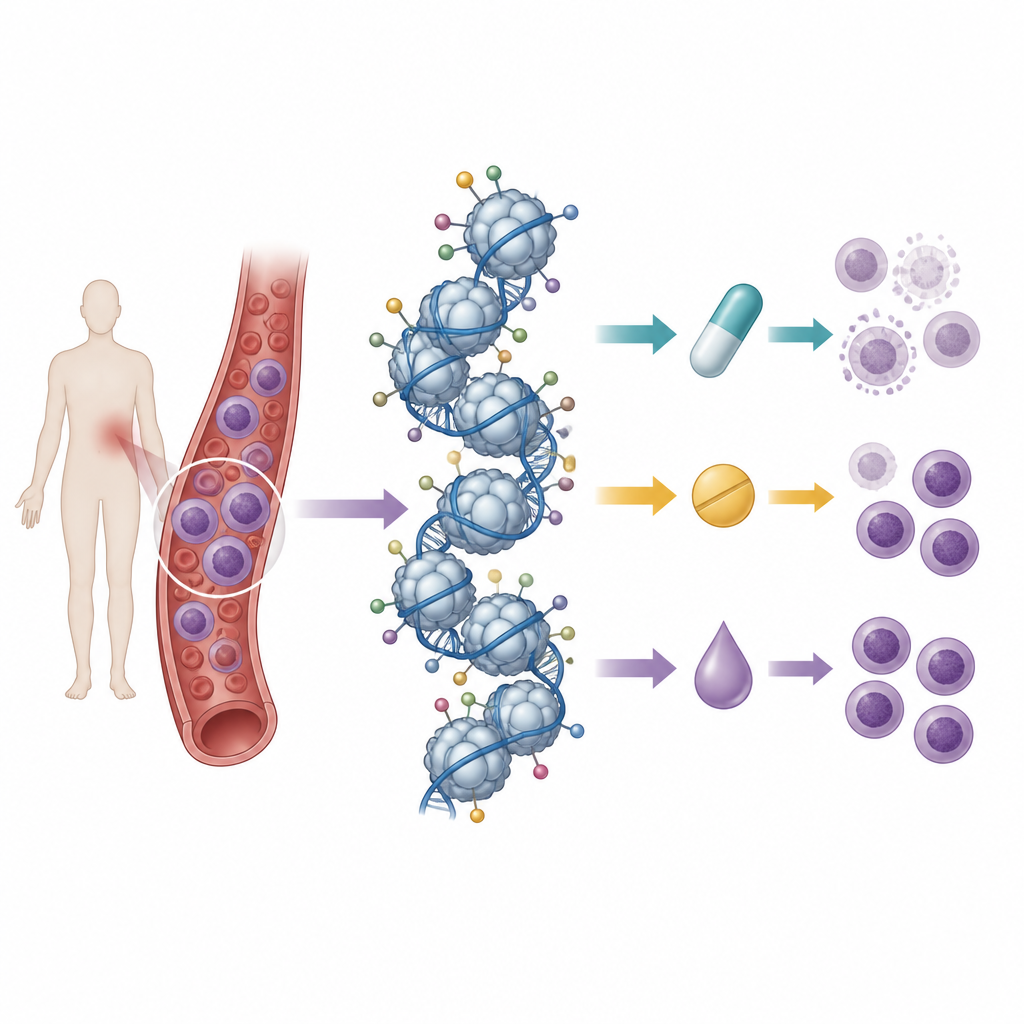
Reading many histone marks at once
The researchers built on a previous effort in which they had mapped the baseline pattern of histone tags in 21 T cell leukemia cell lines using mass spectrometry, a technique that can measure hundreds of chemical marks at once. They then treated these cell lines with nine drugs: three standard chemotherapy anthracyclines, three drugs that block histone deacetylases, and three that inhibit DNA methylation enzymes. By comparing cell survival after treatment with the starting histone patterns, they looked for “signatures” of tags that tracked with sensitivity or resistance to each drug.
What the cell lines revealed
In cultured cells, distinct histone signatures did emerge. For anthracycline drugs, certain repressive marks, such as one called H3K27me3, tended to be higher in more sensitive lines, while other marks, especially several dimethyl groups, were linked to poorer response. For drugs that target DNA methylation, high levels of some dimethyl marks again pointed to resistance, whereas acetyl marks on histone tails were associated with better response. Histone deacetylase inhibitors showed weaker separation between sensitive and resistant cell lines, but some patterns still appeared, including specific marks enriched in either the least or most responsive cells. Overall, these findings suggested that the combined landscape of histone tags, rather than any single mark, carries information about how leukemia cells respond to epigenetic drugs.
Putting a single mark to the test
Because earlier work had suggested that aclarubicin works especially well in cells rich in the H3K27me3 mark, the team directly tested this idea. They used another drug to block the enzyme that writes this mark and successfully lowered H3K27me3 in several T cell leukemia lines. Surprisingly, this did not change how sensitive the cells were to aclarubicin. They also observed cell lines that naturally had high H3K27me3 yet were resistant. This showed that even when a single histone tag tracks with drug response in some settings, it is not sufficient on its own to explain sensitivity.
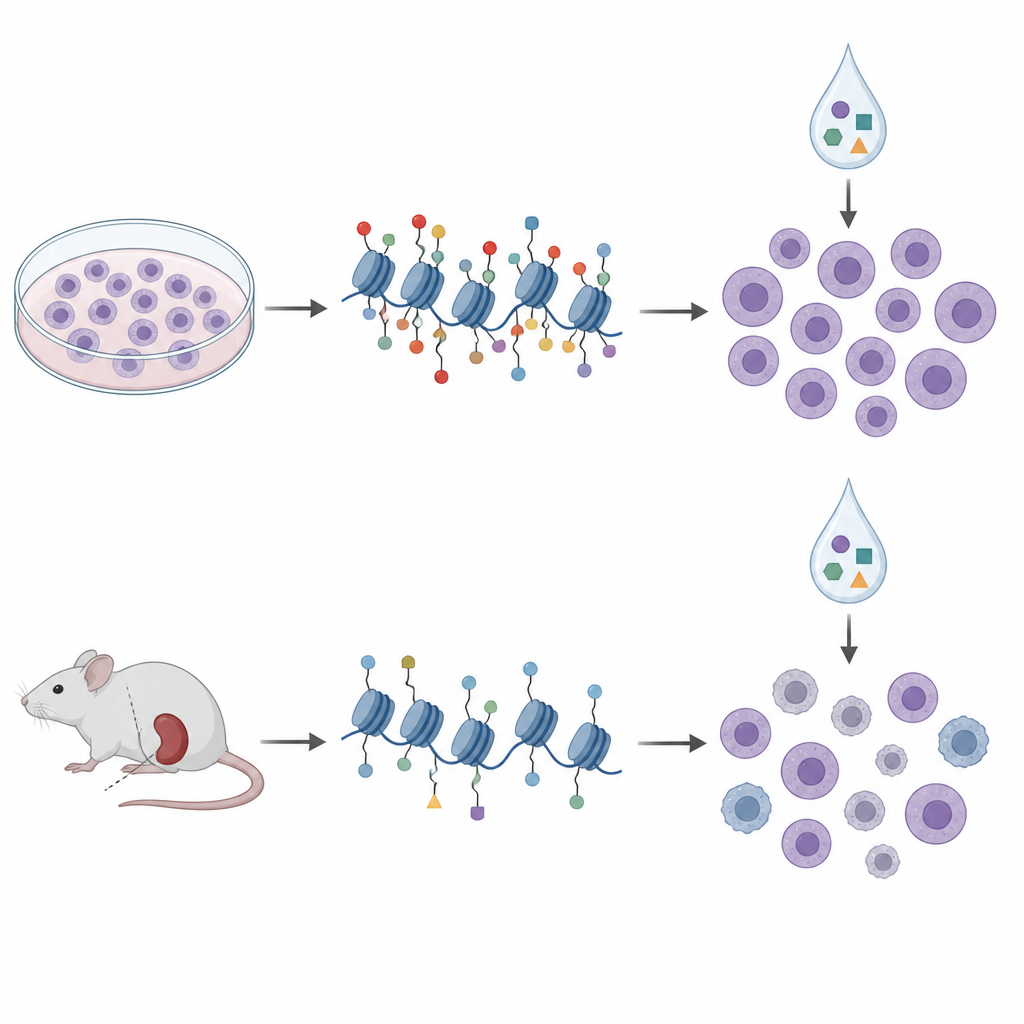
From dishes to mouse models
To move closer to the clinic, the researchers turned to patient-derived xenografts, in which leukemia cells from patients grow in immune-deficient mice. They profiled histone tags and DNA methylation in cells from ten such models and treated them with a similar panel of epigenetic drugs outside the body. In these samples, histone patterns grouped into four clusters that largely mirrored known genetic subtypes and broader DNA methylation states, suggesting that histone profiling captures meaningful disease biology. But when they again compared histone marks to drug sensitivity, the signatures from cell lines did not carry over. In fact, some relationships reversed direction; the same H3K27me3 mark that hinted at aclarubicin sensitivity in dishes was linked to lower sensitivity in the mouse-derived cells.
A shifting network rather than a fixed code
To understand this mismatch, the team studied how different histone marks rise and fall together across the models. In patient-derived samples, many marks moved in tight coordination, forming a dense network of positive correlations, while in cell lines this network was looser and more fragmented. Certain marks sat at the hubs of these networks in one system but not the other. These patterns support the idea that histone tags operate as part of a context-dependent language, where the meaning of any one tag depends on its neighbors and the cellular environment. Long-term growth in a dish seems to rewire this language in ways that change how histone patterns relate to drug response.
What this means for future leukemia care
This work outlines a framework for using mass spectrometry to read the rich landscape of histone marks in T cell leukemia and link them to how cells handle epigenetic drugs. It also delivers a cautionary message: results from standard cell lines may not reliably predict responses in more realistic patient-derived models, let alone in people. For histone-based biomarkers to guide personalized treatments, researchers will need to embrace this complexity, focus on combinations of marks, and carefully choose disease models that reflect the biology of patients as closely as possible.
Citation: Corveleyn, L., Provez, L., Satilmis, O. et al. Profiling histone post-translational modifications to identify signatures of epigenetic drug response in T-cell acute lymphoblastic leukemia. Sci Rep 16, 15029 (2026). https://doi.org/10.1038/s41598-026-44665-4
Keywords: T cell acute lymphoblastic leukemia, epigenetic drugs, histone modifications, patient-derived xenografts, drug response biomarkers