Clear Sky Science · en
Exploring molecular mechanisms of aminoglycoside resistance in Escherichia coli MG1655 using the antibiotic resistance growth plate
Why fast-changing germs matter to everyone
Antibiotics have saved countless lives, but bacteria are steadily learning how to dodge them. This paper explores how ordinary gut bacteria, Escherichia coli, can evolve resistance to a widely used antibiotic, gentamicin, in just a couple of days. By watching resistance unfold on a specially designed growth plate and then probing the bacteria’s DNA and molecular structures, the researchers uncover new details about how these microbes outsmart the drug. Their findings help explain why resistance can arise so quickly, and point to tools that could forecast which treatments are likely to fail.
A new plate to watch evolution in action
To make resistance visible as it emerged, the team built what they call the Antibiotic Resistance Growth Plate (ARGP). Imagine a petri dish divided into three circular bands: no drug in the center, a moderate dose in the middle ring, and a very high dose around the edge. They seeded the center with a standard, drug-sensitive strain of E. coli and let the bacteria swim outward through a thin, semi-solid layer. As the microbes spread, they encountered higher and higher gentamicin levels. Most were stopped at the borders between zones, but every so often tiny offshoots managed to cross into the next ring and keep growing. Within just two to three days, some lineages were thriving in antibiotic concentrations ten times higher than the minimum needed to stop the original strain. 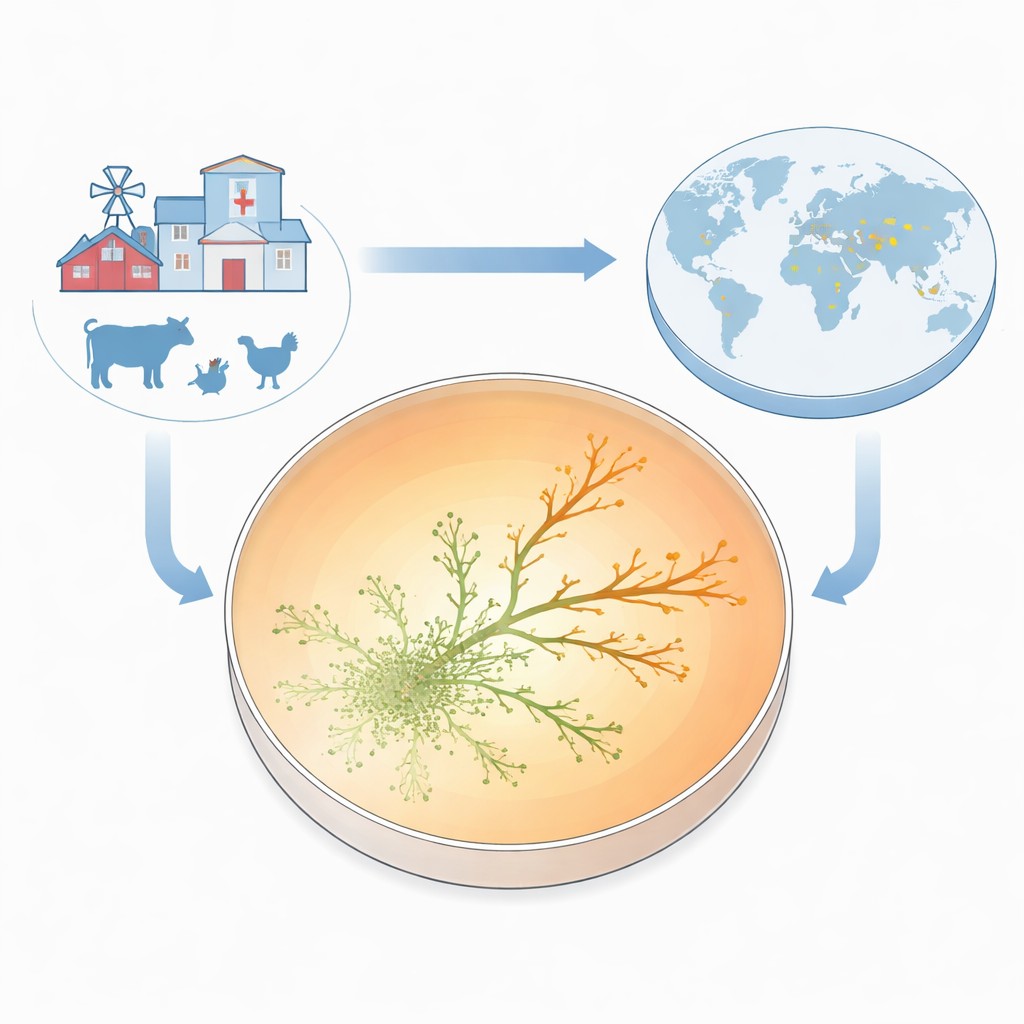
Tracking the genetic changes behind survival
Seeing resistant colonies appear was only the start. The scientists collected cells from these hardy outgrowths and compared their entire DNA sequence to that of the original strain. After carefully filtering out artefacts and contaminating fragments, they focused on changes in genes known to be involved in protein production inside the cell. They found mutations in parts of the genetic code that build the cell’s protein-making machinery: several copies of the 16S ribosomal RNA gene, and a gene called fusA, which encodes a key helper protein known as EF-G. Both targets make sense because gentamicin works by latching onto the bacterial ribosome, the complex that reads genetic instructions and stitches together new proteins. Any tweak that alters this machinery without breaking it entirely has the potential to blunt the drug’s effect.
How a single hinge change weakens the drug
The most striking alteration was a single-letter change in the fusA gene that swaps one amino acid—proline—for another—threonine—at position 610 in the EF-G protein. EF-G acts like a movable arm that helps shift transfer RNAs and messenger RNA through the ribosome as each new protein is built. Using computer models of EF-G’s three-dimensional structure, the team showed that this position sits at a hinge between two of EF-G’s domains, where the protein bends and swivels as it pushes the ribosome along. The proline at this hinge appears to be unusually well conserved across many related bacteria, hinting at its importance. When it is replaced, the overall flexibility and compactness of EF-G change, subtly reshaping how the protein fits against the ribosome. Docking simulations that virtually place gentamicin onto ribosome–EF-G complexes suggest that this tiny structural shift alters the geometry of the drug’s binding region. In the mutated form, gentamicin can no longer trap the ribosome in the vulnerable stage where it normally blocks movement.
Small shifts in RNA structure with big consequences
Mutations also arose in two of the multiple 16S rRNA gene copies that help form the smaller half of the ribosome. By mapping these changes onto known RNA structures, the authors found that all of them clustered in the “5′ body” region, near where gentamicin and a nearby ribosomal protein normally dock. Detailed computer predictions showed that the overall RNA fold was largely preserved, but local loops and stems near the mutated bases were subtly rearranged. These small structural tweaks are enough to change how the RNA interacts with both ribosomal proteins and the drug, potentially nudging critical bases away from the precise positions gentamicin needs for tight binding. When combined with the EF-G hinge mutation, the result is a ribosome that continues to read genetic instructions even in the presence of high drug levels. 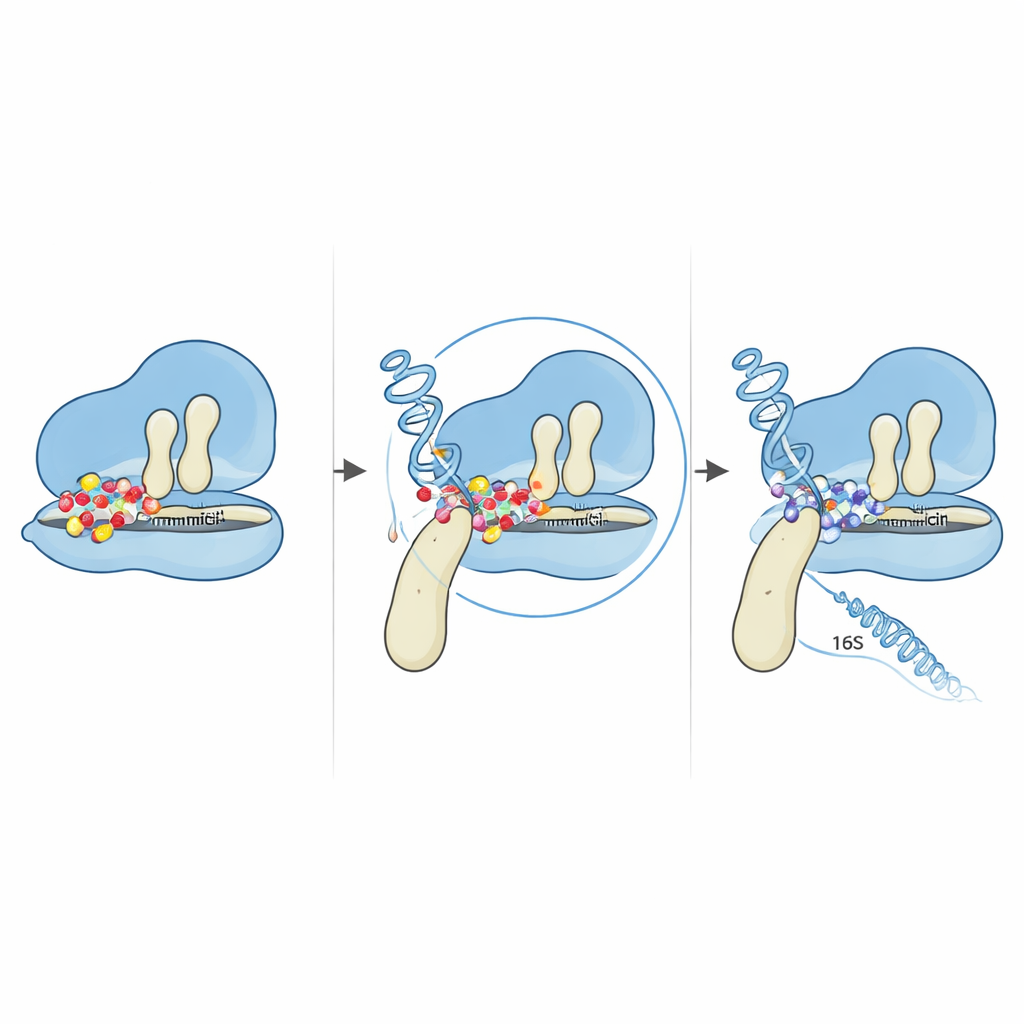
What this means for tackling resistance
Taken together, the work shows that E. coli can rapidly assemble a highly specific defense against gentamicin by modifying both the drug’s docking site on ribosomal RNA and a moving part of the protein engine that drives translation. These changes do not destroy the protein factory; they fine‑tune it just enough that the antibiotic can no longer jam the works. The ARGP system provides a simple, visual way to generate and study such resistance pathways under controlled conditions, and could be adapted to other drugs and pathogens. For non-specialists, the key message is that resistance is not a vague, slow trend—it can arise quickly through precise molecular edits. Understanding those edits in detail is essential if we are to design new antibiotics and treatment strategies that stay one step ahead of evolving bacteria.
Citation: Cullen, L., Eldridge, C., Jones, B. et al. Exploring molecular mechanisms of aminoglycoside resistance in Escherichia coli MG1655 using the antibiotic resistance growth plate. Sci Rep 16, 11958 (2026). https://doi.org/10.1038/s41598-026-41386-6
Keywords: antibiotic resistance, gentamicin, Escherichia coli, ribosome, experimental evolution