Clear Sky Science · en
Complexoform-restricted covalent TRMT112 ligands that allosterically agonize METTL5
Targeting One Player in a Crowded Cell
Inside every human cell, thousands of different protein machines jostle for space, switching on and off to control everything from energy use to brain wiring. Many of these machines are built from several protein parts that snap together in different combinations, creating families of look‑alike complexes. This paper shows how chemists can build small molecules that latch onto just one specific version of such a complex and, instead of shutting it down, actually make it work harder. The work focuses on a protein pair that helps tune how ribosomes read genetic information, offering a new way to adjust protein production with great precision.
Why Protein Mix‑and‑Match Matters
Our roughly 20,000 genes give rise to far more than 20,000 distinct protein entities. After RNA splicing, chemical modifications, and assembly into multi‑protein machines, each gene can spawn many "proteoforms." One important category is "complexoforms"—different combinations of proteins that form related but distinct complexes. A small adaptor protein called TRMT112 is a striking example. It acts as a hub, binding several different methyltransferase enzymes that attach tiny chemical tags to RNA, DNA, or proteins. Because TRMT112 partners with many enzymes, turning it off genetically or with a blunt drug would scramble multiple pathways at once. The authors instead asked: can we find a compound that only binds a single TRMT112 partnership and changes the behavior of just that one enzyme?
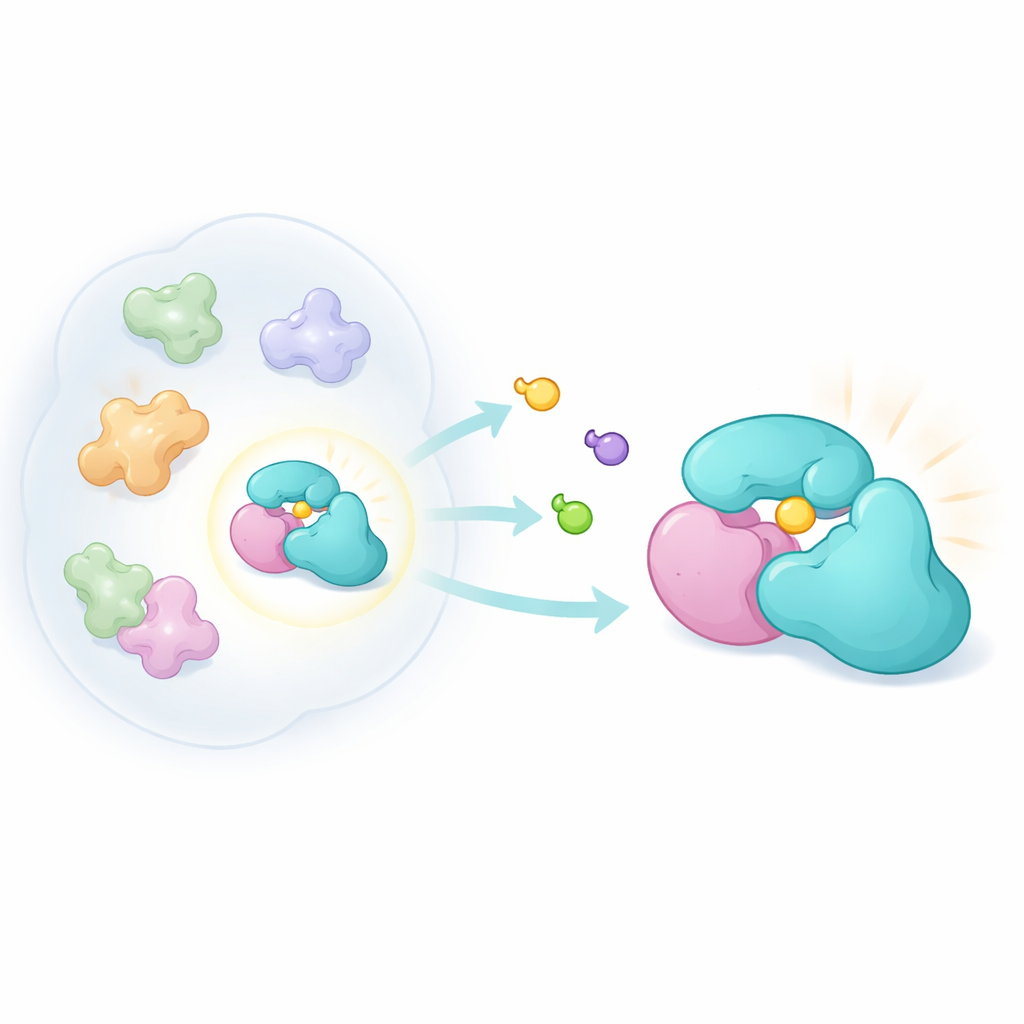
Designing Subtle Chemical Probes
To explore this idea, the researchers built a new family of compact, three‑dimensional molecules called bicyclopyrrolidine acrylamides. These compounds are designed to react covalently with cysteine, a sulfur‑containing amino acid that often sits in reactive pockets on proteins. Using a chemoproteomic method known as activity‑based protein profiling, they exposed living cancer cells to pairs of mirror‑image versions of these compounds and then used mass spectrometry to see which proteins were tagged. By comparing how strongly each mirror image bound, they could spot highly shape‑dependent interactions. Despite being less generally reactive than earlier probe families, the new compounds hit a distinct set of proteins, including TRMT112.
Finding a Single Favored Partnership
Closer analysis revealed an odd mismatch. One assay that watches whole proteins suggested that the probes almost completely blocked TRMT112 from being labeled by a standard tagging reagent. Yet a second, more direct assay that looks at individual cysteine sites showed only a modest effect on one particular cysteine in TRMT112. This hinted that the compounds were recognizing only a fraction of all TRMT112 molecules—the fraction bound to a particular partner. By separating cell extracts by size and by co‑expressing different known TRMT112 partners, the team showed that probe binding appeared only when TRMT112 was paired with one enzyme, METTL5, which modifies a specific adenine base in the cell’s small ribosomal RNA. Knocking out METTL5 erased the interaction, and varying METTL5 levels across cell lines changed how much TRMT112 was engaged, confirming that the probe recognizes a TRMT112:METTL5 complexoform.
Seeing and Tuning the Molecular Interface
Having pinpointed this partnership, the authors refined their molecules to bind more tightly and selectively. An optimized compound, FWG‑33B, reacted with a single cysteine on TRMT112 located right at the interface with METTL5. X‑ray crystallography of the complex showed FWG‑33B wedged into a pocket formed jointly by both proteins, far from METTL5’s active site but close enough to trigger a subtle reshaping of a flexible loop near the enzyme’s cofactor‑binding region. This composite pocket exists only in the TRMT112:METTL5 complex and not in similar complexes TRMT112 forms with other enzymes. Biochemical assays with synthetic RNA fragments revealed the functional outcome: after covalent binding of FWG‑33B, METTL5 bound its RNA substrate more readily and methylated it about twice as efficiently, while a control mutation that removed the targeted cysteine abolished this boost.
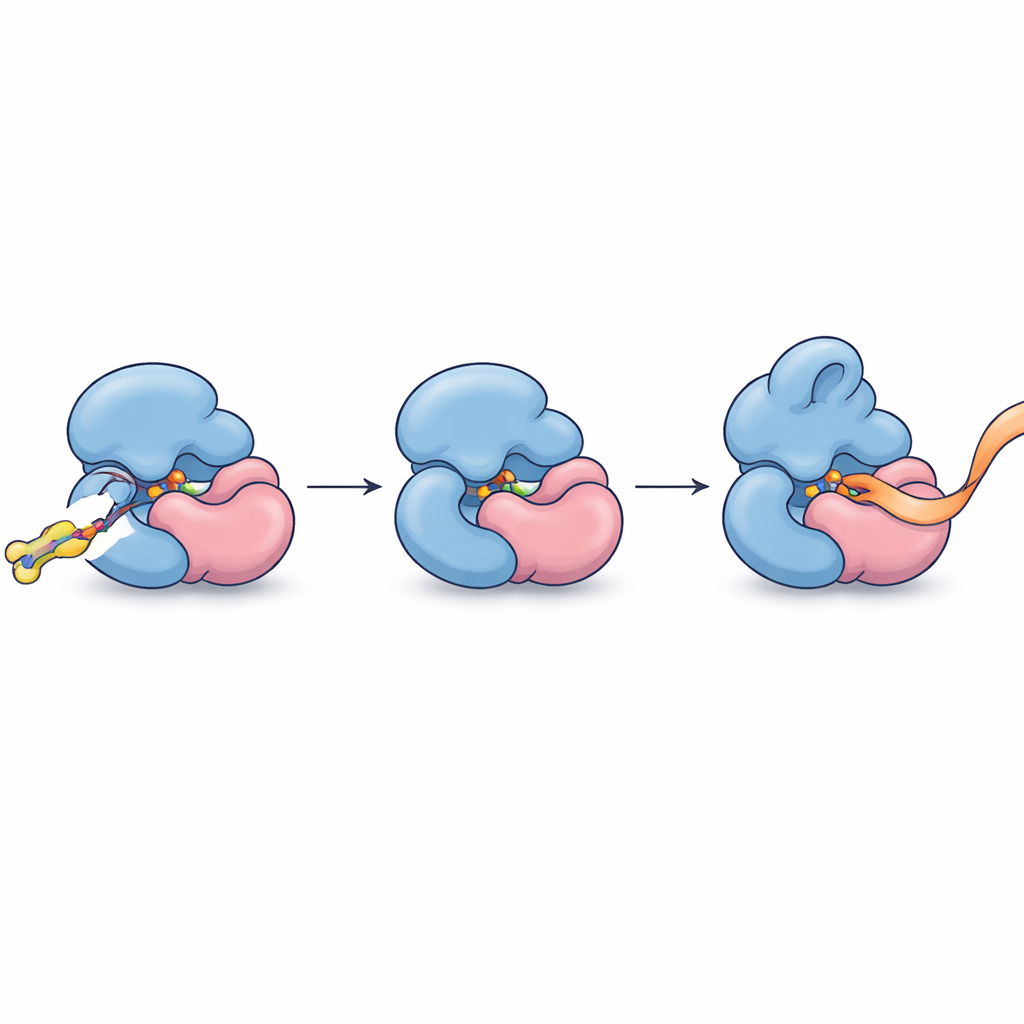
Turning Precision Binding into Precision Control
In everyday terms, the researchers have found a way to slip a tiny molecular wedge into the seam between two protein parts in order to subtly tighten and re‑shape their grip on RNA, making one specific methylating enzyme work faster without disturbing its cousins. This demonstrates that even highly connected adaptor proteins, once thought too blunt a target, can be addressed at the level of individual complexes using covalent chemistry and proteome‑wide profiling. Beyond providing the first allosteric chemical agonists for METTL5—an enzyme increasingly linked to brain development, metabolism, and cancer—the study outlines a general blueprint for discovering small molecules that recognize and tune single members of crowded protein families inside their native cellular environment.
Citation: Goetzke, F.W., Bernard, S.M., Ju, CW. et al. Complexoform-restricted covalent TRMT112 ligands that allosterically agonize METTL5. Nat Chem Biol 22, 770–782 (2026). https://doi.org/10.1038/s41589-025-02099-5
Keywords: protein complexes, chemical probes, RNA methylation, allosteric modulation, chemoproteomics