Clear Sky Science · en
Mechanism of beta-arrestin 1 mediated Src activation via Src SH3 domain revealed by cryo-electron microscopy
How Cells Turn Signals into Action
Our cells constantly sense hormones and neurotransmitters through receptors on their surface and must decide when to respond, when to stop, and which internal pathways to switch on. This study looks at two key players in that decision-making process—beta-arrestin 1 and the enzyme Src—to reveal, in atomic detail, how one protein physically flips the "on" switch of another. Understanding this choreography helps explain how everyday signaling works and may shed light on what goes wrong in diseases such as cancer, where Src is often overactive.
A Traffic Cop That Is Also a Switch
G protein–coupled receptors (GPCRs) are a huge family of cell-surface sensors that control processes from vision to heart rate. Beta-arrestins were long thought of mainly as cellular traffic cops: they latch onto activated GPCRs, shut down further signaling, and guide receptors inside the cell. Over the last decade, however, beta-arrestins have emerged as signal carriers in their own right, able to connect GPCRs to many downstream enzymes. One of the most important partners is Src, a tyrosine kinase that can drive cell growth, movement, and survival. Yet the precise way beta-arrestin 1 (β-arrestin 1, or βarr1) talks to Src—and actually turns it on—has remained a mystery because the interaction is fleeting and relatively weak.
Two Handshakes on One Protein
Using cryo–electron microscopy, which can visualize protein assemblies at near-atomic resolution, the authors captured βarr1 bound to the SH3 domain of Src, a small docking module found in many signaling proteins. They discovered that βarr1 does not rely on a single contact point. Instead, it offers two separate handshakes on different parts of its structure. One site sits in the N-terminal region of βarr1 and contains a short stretch rich in prolines—amino acids that often engage SH3 domains. The second site, in a region called the central crest, uses a different mix of amino acids to land on the same aromatic surface of the SH3 domain. Biophysical measurements and hydrogen–deuterium exchange experiments confirm that both areas of βarr1 become protected when SH3 is present, even without engineered cross-links, showing that these dual contacts occur under realistic conditions.
Unlocking an Autoinhibited Enzyme
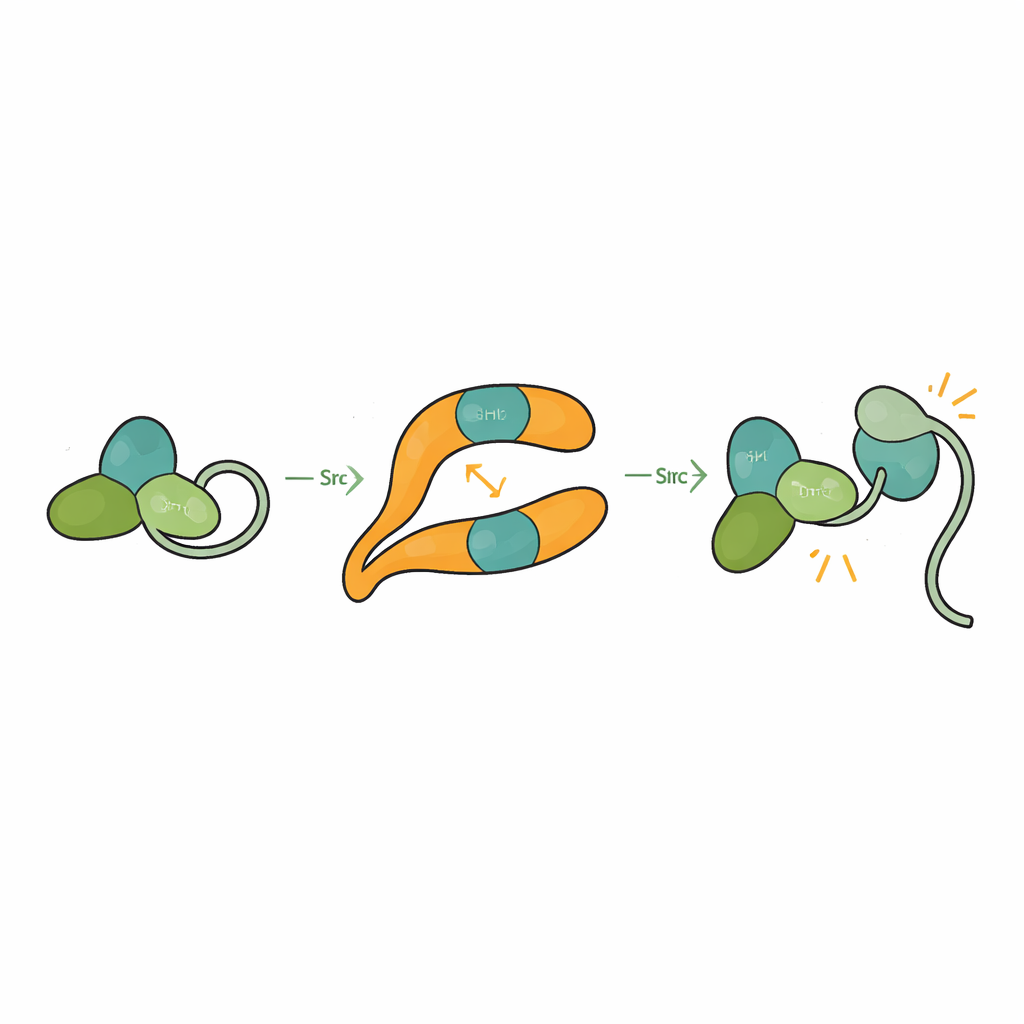
Src is normally held in a self-clamped, or autoinhibited, shape in which its SH3, SH2, and catalytic domains pack against each other and a tail segment tethers the structure closed. This prevents accidental activation that could damage the cell. By solving the structure of βarr1 bound to a three-domain form of Src, the researchers saw that when SH3 engages the central crest site on βarr1, it can no longer clasp the internal linker that helps keep Src shut. The catalytic domain becomes more mobile and the overall shape loosens, consistent with an active state. Biochemical assays show that βarr1 preferentially binds Src when its tail is not phosphorylated, a more flexible form that momentarily exposes SH3. Once SH3 latches onto βarr1, the clamp that keeps Src quiet is pried open, allowing Src to autophosphorylate and fully activate.
A Flexible Hub for Signal Routing
The study also reveals that βarr1 itself is not rigid. When SH3 binds, key strands and loops within βarr1 shift by several angstroms, especially around the central crest. These subtle motions may alter how tightly βarr1 grips GPCRs. Experiments using fluorescently labeled βarr1 show that an excess of SH3 can weaken the way βarr1 inserts part of itself into the receptor core, a configuration thought to be important for shutting down G protein signaling. In living cells, overexpressing Src prolongs the rise in a second messenger called cAMP after activating a beta-adrenergic receptor, indicating delayed desensitization. Together, these findings suggest that high local Src levels can tilt GPCR–βarr1 assemblies away from full "off" mode and allow G proteins to keep signaling longer.
Why This Matters for Health and Therapy
By directly visualizing how βarr1 grabs Src at two sites and pulls it out of its self-inhibited pose, this work establishes βarr1 as an active regulator, not just a passive scaffold. It shows that a single adaptor protein can combine multiple, relatively weak contacts into a robust and tunable control over a powerful enzyme. Because Src is frequently overexpressed or misregulated in cancers, understanding this activation route could inspire strategies that selectively disrupt βarr1–Src communication without shutting down other Src functions. More broadly, the dual-site recognition uncovered here may be a general blueprint for how beta-arrestins engage many of their partners, offering fresh angles for designing drugs that bias GPCR signaling toward beneficial pathways while avoiding harmful ones.
Citation: Pakharukova, N., Thomas, B.N., Bansia, H. et al. Mechanism of beta-arrestin 1 mediated Src activation via Src SH3 domain revealed by cryo-electron microscopy. Nat Commun 17, 2973 (2026). https://doi.org/10.1038/s41467-026-69884-1
Keywords: beta-arrestin, Src kinase, GPCR signaling, cryo-electron microscopy, cell signaling