Clear Sky Science · de
Mechanismus der beta‑Arrestin‑1‑vermittelten Src‑Aktivierung über die Src‑SH3‑Domäne aufgeklärt durch Kryo‑Elektronenmikroskopie
Wie Zellen Signale in Aktion verwandeln
Unsere Zellen nehmen ständig Hormone und Neurotransmitter über Rezeptoren an ihrer Oberfläche wahr und müssen entscheiden, wann sie reagieren, wann sie stoppen und welche inneren Signalwege sie einschalten. Diese Studie untersucht zwei Schlüsselfiguren in diesem Entscheidungsprozess – beta‑Arrestin 1 und das Enzym Src – und zeigt auf atomarer Ebene, wie ein Protein physikalisch den „An“-Schalter eines anderen umlegt. Dieses Zusammenspiel erklärt, wie alltägliche Signalübertragung funktioniert, und kann Aufschluss darüber geben, was bei Krankheiten wie Krebs schiefläuft, in denen Src häufig überaktiv ist.
Ein Verkehrspolizist, der auch ein Schalter ist
G‑Protein‑gekoppelte Rezeptoren (GPCRs) sind eine riesige Familie von Sensorsystemen an der Zelloberfläche, die Prozesse von der Sehkraft bis zur Herzfrequenz steuern. Lange galten Beta‑Arrestine hauptsächlich als zelluläre Verkehrspolizisten: Sie binden an aktivierte GPCRs, schalten weitere Signale ab und lenken Rezeptoren in die Zelle. In den letzten zehn Jahren haben sich Beta‑Arrestine jedoch als eigenständige Signalträger herauskristallisiert, die GPCRs mit vielen nachgeschalteten Enzymen verbinden können. Einer der wichtigsten Partner ist Src, eine Tyrosinkinase, die Zellwachstum, Bewegung und Überleben antreiben kann. Wie beta‑Arrestin 1 (β‑arrestin 1 bzw. βarr1) genau mit Src kommuniziert und es tatsächlich aktiviert, war jedoch lange rätselhaft, weil die Wechselwirkung kurzlebig und vergleichsweise schwach ist.
Zwei Händedrücke an einem Protein
Mithilfe der Kryo‑Elektronenmikroskopie, die Proteinverbände in nahezu atomarer Auflösung sichtbar machen kann, erfassten die Autoren βarr1 gebunden an die SH3‑Domäne von Src, ein kleines Andockmodul, das in vielen Signalmolekülen vorkommt. Sie entdeckten, dass βarr1 sich nicht auf einen einzigen Kontaktpunkt verlässt. Stattdessen bietet es zwei getrennte „Händedrücke“ an verschiedenen Stellen seiner Struktur. Eine Stelle liegt im N‑terminalen Bereich von βarr1 und enthält eine kurze, prolinreiche Sequenz – Aminosäuren, die oft SH3‑Domänen ansprechen. Die zweite Stelle befindet sich in einer Region, die als zentraler Kamm bezeichnet wird, und verwendet eine andere Mischung von Aminosäuren, um auf derselben aromatischen Fläche der SH3‑Domäne zu landen. Biophysikalische Messungen und Wasser‑Deuterium‑Austausch‑Experimente bestätigen, dass beide Bereiche von βarr1 geschützt werden, wenn SH3 präsent ist – selbst ohne konstruierte Quervernetzungen – und zeigen damit, dass diese doppelten Kontakte unter realistischen Bedingungen auftreten.
Eine autoinhibierte Kinase entriegeln
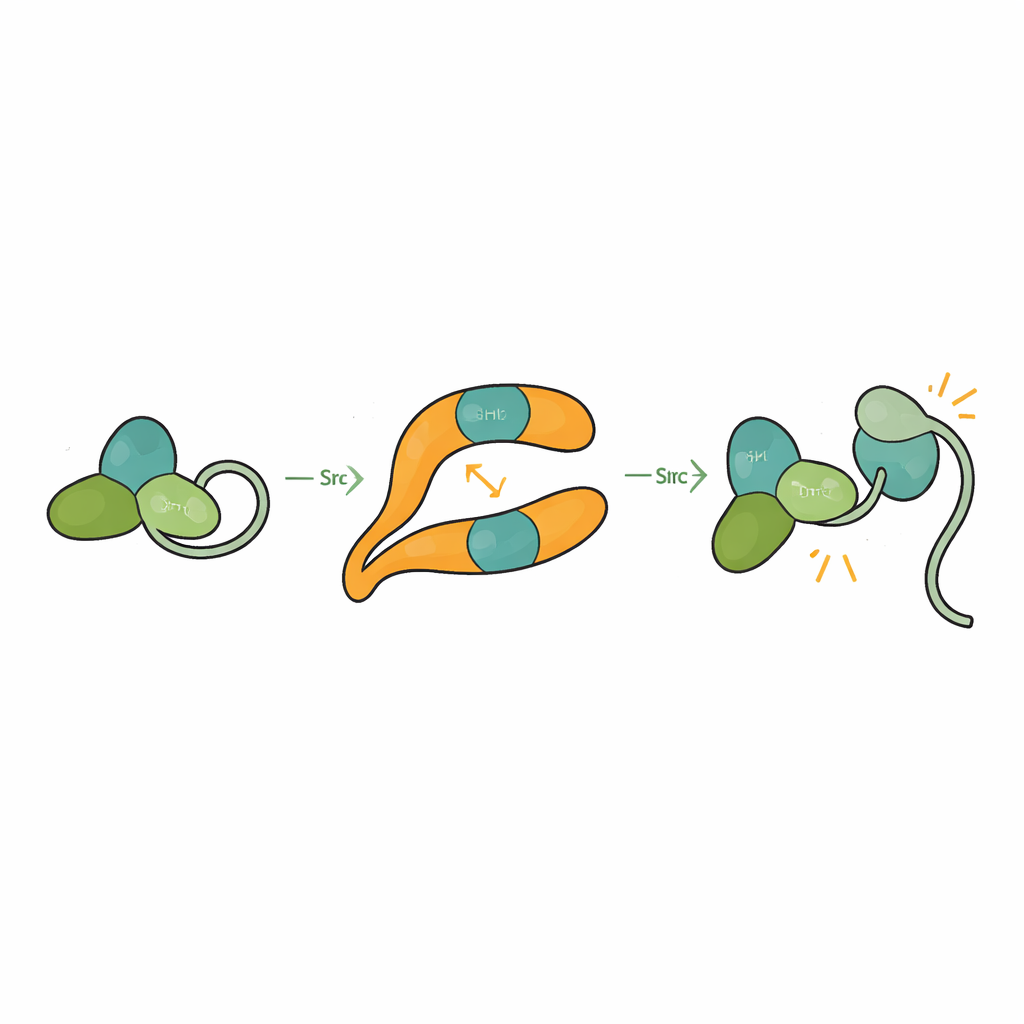
Src liegt normalerweise in einer selbstgeklammerten, also autoinhibierten Konformation vor, in der seine SH3‑, SH2‑ und katalytischen Domänen aneinander anliegen und ein C‑terminales Schwanzsegment die Struktur verschlossen hält. Das verhindert eine versehentliche Aktivierung, die der Zelle schaden könnte. Durch die Lösung der Struktur von βarr1 gebunden an eine Drei‑Domänen‑Form von Src beobachteten die Forschenden, dass, wenn SH3 die zentrale Kamm‑Stelle auf βarr1 einnimmt, es nicht mehr in der Lage ist, den internen Linker zu umschlingen, der Src geschlossen hält. Die katalytische Domäne wird beweglicher und die Gesamtform lockert sich, was mit einem aktiven Zustand übereinstimmt. Biochemische Assays zeigen, dass βarr1 bevorzugt an Src bindet, wenn dessen Schwanz nicht phosphoryliert ist – eine flexiblere Form, die zeitweilig die SH3‑Domäne freilegt. Sobald SH3 an βarr1 anschlägt, wird die Klammer, die Src ruhig hält, aufgebrochen, sodass Src autophosphorylieren und vollständig aktiviert werden kann.
Ein flexibler Knotenpunkt für Signalweiterleitung
Die Studie zeigt außerdem, dass βarr1 selbst nicht starr ist. Wenn SH3 bindet, verschieben sich entscheidende Stränge und Schleifen innerhalb von βarr1 um mehrere Ångström, besonders rund um den zentralen Kamm. Diese subtilen Bewegungen könnten die Bindungsstärke von βarr1 an GPCRs verändern. Experimente mit fluoreszenzmarkiertem βarr1 zeigen, dass ein Überschuss an SH3 die Art und Weise abschwächen kann, wie βarr1 einen Teil von sich in den Rezeptorkern einfügt – eine Konfiguration, die als wichtig für das Abschalten der G‑Protein‑Signalgebung gilt. In lebenden Zellen verlängert die Überexpression von Src den Anstieg des zweiten Botenstoffs cAMP nach Aktivierung eines beta‑adrenergen Rezeptors, was auf verzögerte Desensitivierung hindeutet. Zusammen deuten diese Befunde darauf hin, dass hohe lokale Src‑Spiegel GPCR–βarr1‑Assemblies von einem vollständigen „Aus“-Zustand wegkippen lassen und es den G‑Proteinen ermöglichen, länger zu signalisieren.
Warum das für Gesundheit und Therapie wichtig ist
Indem diese Arbeit direkt zeigt, wie βarr1 Src an zwei Stellen greift und aus seiner selbst‑inhibierten Pose herauszieht, etabliert sie βarr1 als aktiven Regulator und nicht nur als passives Gerüst. Sie demonstriert, dass ein einzelnes Adapterprotein mehrere, vergleichsweise schwache Kontakte zu einer robusten und einstellbaren Kontrolle über ein leistungsfähiges Enzym kombinieren kann. Da Src bei Krebs häufig überexprimiert oder fehlreguliert ist, könnte das Verständnis dieses Aktivierungswegs Strategien inspirieren, die selektiv die βarr1–Src‑Kommunikation unterbrechen, ohne andere Src‑Funktionen abzuschalten. Allgemeiner könnte die hier entdeckte Doppelstellen‑Erkennung ein allgemeines Prinzip erklären, wie Beta‑Arrestine viele ihrer Partner binden, und neue Ansätze für die Gestaltung von Medikamenten eröffnen, die GPCR‑Signale in nützliche Pfade lenken, während schädliche vermieden werden.
Zitation: Pakharukova, N., Thomas, B.N., Bansia, H. et al. Mechanism of beta-arrestin 1 mediated Src activation via Src SH3 domain revealed by cryo-electron microscopy. Nat Commun 17, 2973 (2026). https://doi.org/10.1038/s41467-026-69884-1
Schlüsselwörter: beta‑Arrestin, Src‑Kinase, GPCR‑Signalübertragung, Kryo‑Elektronenmikroskopie, Zell‑Signalgebung